Frontiers in Physiology
Introduction
A vérszegénység az egyik leggyakoribb betegség világszerte, és több elemzés szerint a vashiány okozta vérszegénység a leggyakoribb forma (review in Camaschella, 2019). Ez a fajta vérszegénység a teljes szervezet vashiányából és abból ered, hogy a csontvelő nem képes biztosítani azt a nagy mennyiségű vasat, amelyet a szövetek oxigénellátásának fenntartásához szükséges megfelelő számú vörösvértest előállításához a csontvelő fogyaszt.
A vas rendelkezésre állását a máj peptidhormonja, a hepcidin szabályozza. A szervezet vasszintjének növekedése hepcidin termelődését okozza, amely a keringésbe kerül, és receptorára, a ferroportinra hat, amely egy transzmembrán vaskivitelező fehérje, amely az enterocitákon, makrofágokon és hepatocitákon nagymértékben expresszálódik. A hepcidin csökkenti a vas bejutását a plazmába a felszívódó nyombélsejtekből és a vasat újrahasznosító makrofágokból azáltal, hogy gátolja a vaskivitelt (Aschemeyer és mtsai., 2018) és lebontja a vaskivitelező ferroportint (Németh és mtsai., 2004). A plazma vas és a szisztémás vashomeosztázis szabályozásával a hepcidin/ferroportin tengely erősen befolyásolja az eritropoiesist, ezáltal az anémia lehetséges kialakulását.
A vas-eritropoiesis kapcsolat
A vörösvértestek termelődésének folyamata a keringő vas mintegy 80%-át az érő eritroblasztok hemoglobinszintéziséhez használja fel. A vas nagy részét (20-25 mg/nap) a makrofágok újrahasznosítják, míg korlátozott mennyiség (napi 1-2 mg) a bélrendszeri felszívódásból származik. Az eritropoetin (EPO) nevű vesehormon szabályozza az eritroid progenitorok, különösen a CFU-e és kisebb mértékben a BFU-e proliferációját, valamint a terminális eritropoiesis korai szakaszát, míg a vasszükséglet a proeritroblastokból retikulocitává válás késői differenciálódási szakaszában a hemoglobinba beépülő hem szintéziséhez megnő (Muckenthaler et al., 2017).
A hepcidin szabályozásához a BMP-SMAD útvonalat aktiváló csontmorfogenetikus fehérjéket (BMP-ket) termelő máj endothelialis sinusoidális sejtek (LSEC) és a hepcidint termelő és felszabadító hepatociták közötti keresztkapcsolat szükséges (Babitt et al., 2006; Rausa et al., 2015). A BMP6 és BMP2 a legfontosabb BMP-k, amelyek felszabályozzák a hepcidint, míg a BMP6 expressziója vasfüggő (Andriopoulos et al., 2009; Meynard et al., 2009), a BMP2 kevésbé tűnik vasfüggőnek (Canali et al., 2017; Koch et al., 2017).
A hepcidin szintje alacsony abszolút vashiányban és vashiányos anémiában. Ezekben az állapotokban a vasraktárak kimerülnek, és a BMP-SMAD jelátvitel több szinten kikapcsol. Először is, a BMP6 expressziója elnyomódik; ezután a TMPRSS6, a BMP társreceptor hemojuvelint hasító proteáz (Silvestri és mtsai., 2008) aktivitása erősen megnő (Lakhal és mtsai., 2011); harmadszor pedig a hiszton-deacetiláz3 (HDAC3) elnyomja a hepcidin-lokuszt (Pasricha és mtsai., 2017). Vashiányos körülmények között a hepcidin-termelés csökkenése olyan adaptációs mechanizmus, amely megkönnyíti a diétás és farmakológiai vasfelszívódást (Camaschella és Pagani, 2018).
Súlyos vérszegénység esetén az egyidejűleg fennálló hipoxia az EPO fokozott vese-szintézisén és felszabadulásán keresztül serkenti az eritropoézist. Ez a hepcidin transzkripció szuppressziójához vezet az eritroferron (ERFE), az EPO célgénje által, amelyet az eritroblastok termelnek (Kautz és mtsai., 2014), más szövetek által felszabaduló molekulák (pl. PDGF-BB) (Sonnweber és mtsai., 2014), és valószínűleg a transzferrinreceptorok (TFR) oldható komponensei, az sTFR1 (Beguin, 2003) és az sTFR2 (Pagani és mtsai., 2015). A végső cél a kiterjesztett erythropoiesis szükségleteihez elegendő vas biztosítása.
Anemiák kóros hepcidinszintekkel
Anemiák a hepcidinszintek alapján magas és alacsony hepcidinszintű anémiákba sorolhatók. Értelemszerűen a tartósan magas hepcidinszint a vasfelszívódás gátlásával vashiányos anémiát okoz, mivel az eritropoézis vasellátása csökken. Ezzel szemben a nem hatékony erythropoiesis jellemzi az úgynevezett vasterheléses anémiákat, amelyek alacsony hepcidinszinttel és vastúlterheléssel járnak. Az anémiák e két csoportja ellentétes patofiziológiai mechanizmusok eredménye (1. ábra). Az első csoportban a vérszegénység a hepcidin által a vas felszívódására és újrahasznosítására kifejtett gátló hatásnak köszönhető, amely szisztémás vashiányhoz vezet; a második csoportban a vérszegénység a hepcidin szuppressziójának köszönhető, amelyet egy kiterjedt kóros erythropoiesis okoz (Camaschella és Nai, 2016).
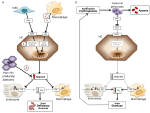
1. ábra. A magas (bal oldali panel) és alacsony hepcidinszintű (jobb oldali panel) anémiák mechanizmusainak sematikus ábrázolása. Panel (A). A magas hepcidinszinttel járó anémia molekuláris patogenezise. LSEC, csontmorfogenetikus fehérjéket (BMP) termelő máj szinuszoidális endotélsejtek; BMPRs, BMP-receptorok; IL6, interleukin 6; HC, hepatociták; HAMP, hepcidin gén. Fe, vas; FPN, ferroportin; 1, IRIDA; 2, gyulladásos vérszegénység; 3, hepcidin-termelő adenóma. Panel (B). A hepcidin-variáció molekuláris patogenezise az ineffektív erythropoiesis okozta anémiákban. ERFE, eritroferront szekventáló BMP-k. Az ilyen típusú anémiában a hepcidint gátló egyéb mechanizmusok, mint a transzferrin-szaturáció csökkenése és a hypoxia, nem szerepelnek. A részleteket lásd a szövegben.
magas hepcidin-szinttel társuló anémia
Ez a csoport két öröklött ritka rendellenességet (vas-refrakteres vashiányos anémia és a glükózanyagcsere veleszületett hibájából eredő hepcidin-termelő adenomák) és egy szerzett gyakori állapotot: gyulladásos anémia (1. táblázat) foglal magában.
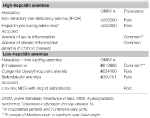
1. táblázat. A hepcidinszintek alapján osztályozott anémiák.
Vasrefrakter vashiányos anémia
A vasrefrakter vashiányos anémia (IRIDA) egy ritka recesszív rendellenesség, amelyet hipokróm mikrocitás anémia, alacsony transzferrin-szaturáció és nem megfelelően normális/magas hepcidinszint jellemez. A TMPRSS6 (Finberg és mtsai., 2008) gén mutációi okozzák, amely a II-es típusú szerin proteázt, a matriptáz-2-t kódolja (Du és mtsai., 2008). A TMPRSS6 mutációi a gén mentén terjednek el, és különböző doméneket, különösen a katalitikus domént érinthetik (De Falco és mtsai., 2014). Ez a májban nagymértékben expresszálódó transzmembrán proteáz a sejtfelszíni BMP társreceptor hemojuvelin hasításával gátolja a hepcidin transzkripcióját, így gyengíti a BMP jelátvitelt és a hepcidin szintézisét (Silvestri és mtsai., 2008). A TMPRSS6 működése alapvető fontosságú vashiányban, hogy lehetővé tegye a fokozott vasfelszívódás kompenzációs mechanizmusát.
Az IRIDA születés óta jelen van, és általában gyermekkorban diagnosztizálják. A klasszikus vashiányhoz képest a vasparaméterek atipikusak, és felvetik a betegség gyanúját. A transzferrin százalékos telítettsége erősen csökkent (kevesebb mint 10%), mint a vashiány más formáiban; azonban a vashiánytól eltérően a szérumferritin szintje normális/emelkedett (Camaschella, 2013; De Falco és mtsai., 2013). Ez a ferritin fokozott felhalmozódását tükrözi a makrofágokban, a magas hepcidinszint miatt, amely raktári vasszekvenciát indukál.
Az IRIDA diagnózisára javasolt tesztek egyike sem fedi le az esetek 100%-át. A genetikai vizsgálat azonosítja a TMPRSS6 mutációkat, amelyek egyes esetekben (nonszensz, frame-shift és splicing mutációk) egyértelműen okozói a betegségnek. Más esetekben, mint a korábban nem bejelentett missense mutációk esetében, funkcionális vizsgálatokra van szükség az ok-okozati összefüggés bizonyításához (Silvestri és mtsai., 2013). Ezek a vizsgálatok azonban alig állnak rendelkezésre. A szérum hepcidinszint általában emelkedett/normális, függetlenül a vashiánytól, és összhangban van a magas/normális ferritinnel. Fontos a gyulladás kizárása a C-reaktív fehérje egyidejű adagolásával.
Egyik-másik refrakter vashiányos fenotípusú betegnél egyetlen TMPRSS6 mutáns allélről számoltak be; itt vita tárgya, hogy IRIDA-nak kell-e tekinteni őket vagy sem. Az állapotok spektruma elképzelhető, amely a homozigóta vagy összetett heterozigóta TMPRSS6 mutációk okozta klasszikus súlyos IRIDA-tól a vashiányra való fokozott fogékonyságig terjed, amelyet egyetlen mutáció/polymorf változás okoz. A klasszikus IRIDA előrejelzésére javasolt egyik megközelítés a hepcidin normalizálása más vasparaméterekre, mint a transzferrin telítettség (Tsat)/log hepcidin vagy Tsat/log ferritin arányok (Donker és mtsai., 2016). Más szerzők szerint a legtöbb súlyos IRIDA fenotípusú betegnél biallelikus TMPRSS6 mutáció áll fenn, és ha nem azonosított, a második allél genetikailag okkult lehet (Heeney és mtsi., 2018). Általánosságban elmondható, hogy az egyetlen alléllal rendelkező alanyok fenotípusa enyhébb, mint a két mutációval rendelkezőké, és jobban reagálnak a vaskezelésre (Donker és mtsai., 2016). Érdekes módon számos TMPRSS6 SNP-ről kimutatták, hogy egyes populációkban (An és mtsai., 2012) és véradókban (Sorensen és mtsai., 2019) vashiányra való fogékonyságot biztosítanak.
Egy 5 éves nőnél, akinél eredetileg atipikus IRIDA genotípust találtak, egy TMPRSS6 (I212T) kauzális és egy (R271Q) csendes mutációt (De Falco és mtsai., 2014), digenikus öröklődést jelentettek. Később Fibrodysplasia ossificans progressiva (FOP) diagnózist kapott, egy ritka domináns rendellenesség, amely az ALK2-t kódoló, mutálódott BMP I. típusú receptor gén, az ACVR1 miatt ektopikus csontképződéssel jár a lágyrészekben (Shore et al., 2006). A kóros ALK2R258S allél konstitutívan aktív, mivel a mutáció a gén glicin-szerin-gazdag doménjét érinti, és a BMP/SMAD útvonalat túlműködővé teszi, mivel nem képes megkötni a specifikus inhibitorát, az FKBP12-t (Pagani et al., 2017).
Ez a ritka eset különösen szemléletes. Először is, mivel az ALK2 glicin-szerin-gazdag domén kölcsönhatásba lép az FKBP12-vel, és a mutáció destabilizálja a kötődést, az FKBP12 korábban nem sejtett szerepét tárta fel a máj ALK2 és hepcidin modulátoraként (Colucci et al., 2017). Másodszor, a csont és a máj BMP I-es típusú receptorainak aktiválása közötti kapcsolat azonosításához vezetett. Harmadszor, az eset megerősíti az intakt TMPRSS6 jelentőségét a máj BMP/SMAD jelátvitelének szabályozásában, mivel más, azonos ACVR1-mutációval és feltehetően normális TMPRSS6-mal rendelkező FOP-betegek között nem azonosítottak IRIDA-t (Pagani és mtsai., 2017). Végül, ez az eset összhangban van azzal az elképzeléssel, hogy a TMPRSS6 haploinsufficiencia nem okozhat klasszikus IRIDA-t.
Az IRIDA optimális kezelése meghatározatlan. Az orális vas hatástalan, mivel nem szívódik fel. A C-vitamin hozzáadása szórványos választ tesz lehetővé. Az intravénás vasadás részleges választ vált ki, általában lassabb ütemben, mint a szerzett vashiányos betegeknél. Az EPO klasszikus esetekben hatástalan (De Falco és mtsai., 2013; Heeney és Finberg, 2014).
A hepcidin-termelő adenomák anémiája
Ez egy rendkívül ritka állapot felnőtt betegeknél, akiket a glikogénraktározási betegség 1a érintett, ami egy recesszív rendellenesség a glükóz-6-foszfatáz hiánya miatt, amely a glikogenolízisben és a glükoneogenezisben egyaránt részt vevő reakciót katalizál. A betegség gyakori veszélyes tünete a hipoglikémia. A jelenlegi kezelés az érintett gyermekek meghosszabbított túlélését eredményezi egészen a felnőttkorig, számos szövődmény, például vérszegénység és májadenomák előfordulása mellett. A vérszegénység mikrocitikus és hipokróm, vashiányos és az orális vaskezelésre refrakter. A vérszegénység a műtéti adenómarezekciót követően visszaállt. Az adenomaszövet hepcidin mRNS tekintetében pozitívnak bizonyult, míg a normális környező szövet hepcidin-szuppressziót mutatott, amint az az ektopikus, ellenőrizetlen hepcidin-termelés miatt várható volt (Weinstein és mtsi., 2002). A betegek hematológiai jellemzői hasonlítanak az IRIDA-ra, mivel a vérszegénység közös mechanizmusa a magas hepcidinszint.
Gyulladásos vérszegénység
A gyulladásos vérszegénység (AI), amelyet korábban krónikus betegségek anémiájaként ismertek, egy mérsékelten normokróm-normocitás anémia, amely szisztémás gyulladás és immunaktiváció körülményei között alakul ki. Számos gyakori betegségben fordul elő, beleértve a krónikus fertőzéseket, autoimmun betegségeket, előrehaladott rákot, krónikus vesebetegséget, pangásos szívelégtelenséget, krónikus obstruktív tüdőbetegséget, az idősek vérszegénységét (legalábbis részben) és a graft versus host betegséget. Az AI világszerte az egyik leggyakoribb anémia, és a kórházi betegeknél a leggyakoribb anémia. Az intenzív osztályokon az akut gyulladás hozzájárul az anémia súlyosságához. Az AI hátterében álló molekuláris mechanizmusok sokrétűek és összetettek. Az olyan citokinek, mint az IL1-β, TNF-α és IL-6 makrofágok általi túltermelődése és a limfociták által termelt INF-γ tompítja az EPO-termelést, károsítja az eritropoézisválaszt, növeli a hepcidinszintet, és aktiválhatja az eritrofagocitózist, különösen az akut formákban (Weiss és Goodnough, 2005; Ganz, 2019).
A hepcidint az IL-6 aktiválja az IL-6 receptoron (IL-6R) és a JAK2-STAT3 szignálon keresztül. A hepcidin teljes aktiválásához aktív BMP-SMAD útvonalra van szükség, mivel a BMP jelátvitel inaktiválása csökkenti a hepcidin szintjét a gyulladás állatmodelljeiben (Theurl és mtsai., 2011). A szisztémás vashomeosztázis deregulációja makrofági vasszekvenciót és csökkent felszívódást és újrahasznosítást okoz, ami a transzferrin alacsony telítettségéhez és az eritropoézis és más szövetek vaskorlátozásához vezet.
Az AI hagyományos kezelése az alapbetegség reverzibilitásán/kontrollján alapul, amikor csak lehetséges. Ha a betegség nem kezelhető, és a vérszegénység enyhe, a kockázatok-előnyök gondos értékelésére van szükség, hogy elkerüljük bármely kezelés mellékhatásait. A patofiziológiai alapú kezelések az eritropoetin-szerű vegyületekre és a vasra korlátozódnak. Az eritropoiesist serkentő szerek (ESA) alkalmazása az eritropoiesis expanziójának indukálásával elnyomja a hepcidint. Ezt a megközelítést széles körben alkalmazzák krónikus vesebetegségben, alacsony kockázatú myelodysplasztikus szindrómákban és kemoterápiában részesülő rákos betegeknél. Gondos klinikai ellenőrzésre van azonban szükség, mert a nagy dózisok kardiovaszkuláris mellékhatásokkal járnak. Az intravénás vas adása enyhítheti az erythropoiesis ESA-függő expanziója által okozott vasszűkületet. A szájon át adott vas általában hatástalan, mivel a magas hepcidinszint ellensúlyozza a bélrendszeri felszívódását. A prolil-hidroxiláz gátlói (hipoxia indukálható faktor, HIF-stabilizátorok) krónikus vesebetegségben kísérleti jelleggel, az endogén EPO növelése céljából. A vörösvérsejt-transzfúzióval történő krónikus kezelés az átmeneti hatás és a mellékhatások miatt nem javasolt; súlyos, refrakter anaemiára korlátozódik (Camaschella, 2019; Weiss és mtsai., 2019).
Az alacsony hepcidinszintekhez társuló anémiák
A “vasterheléses anémiák” jellemzői a nem hatékony erythropoiesis és az alacsony vagy nem megfelelően normális hepcidinszint, következményes vastúlterheléssel.” A prototípus a β-talasszémia, egy genetikai recesszív betegség, amely a β-globin génmutációk miatt vérszegénységet és túlzott α-globinlánc-termelést okoz. Ez utóbbi hemikrómként kicsapódik a csontvelőben, károsítva az érő erythroid prekurzorokat, és hatástalan erythropoiesishez vezet. Ez történik a nem transzfúziófüggő thalassémiában vagy thalassemia intermediában, amelynek eritropoiesisére jellemző az éretlen sejtek túlsúlya, amelyek eritroferront szabadítanak fel a máj hepcidin-expressziójának gátlása érdekében. A hepcidinszint általában nagyobb a transzfúziófüggő thalassaemiában, ahol az endogén ineffektív erythropoiesist legalább részben elnyomják a transzfúziók (Camaschella és mtsi., 2016).
A hepcidin-szupressziót az ERFE gén által kódolt TNF-α családba tartozó, az erythroblastok által EPO-stimuláció hatására szintetizált fokozott citokin, az eritroferron (ERFE) közvetíti (Kautz és mtsi., 2014). Az ERFE a keringésbe kerülve szekvenálja a BMP-ket, különösen a BMP6-ot (Arezes és mtsai., 2018), gyengítve a hepcidin szignalizációt vasra adott válaszként. Emellett a hepcidin lókuszon epigenetikai szuppresszió következik be a hiszton-deacetiláz HDAC3 által (Pasricha és mtsai., 2017). Ha az anémia hipoxiát okoz, más mediátorok, például a PDGF-BB (Sonnweber et al., 2014), amelyet különböző sejttípusok szabadítanak fel, elnyomják a hepcidinszintet.
A hepcidinszint egy speciális mechanizmus révén csökken az alacsony kockázatú, gyűrűs szideroblasztokkal járó myelodysplasiában, amely az SF3B1 spliceoszóma gén mutációi miatt kialakuló klonális rendellenesség. A vas felhalmozódik a mitokondriumokban, ami hatástalan erythropoiesishez és szisztémás vastúlterheléshez vezet. A rendellenesen splicelt, megnyúlt ERFE fehérje a vad típusú ERFE-nél erőteljesebben elnyomja a hepcidint (Bondu et al., 2019) és transzfúzió-független vastöltést okoz.
A hepcidin-függő anémiák célzott terápiái
A korábban tárgyalt anémiákért felelős molekuláris mechanizmusok azonosítása ösztönözte a kutatásokat a jelenlegi tüneti kezelést felváltó célzott terápiák kifejlesztésére (Sebastiani és mtsi., 2016; Crielaard és mtsi., 2017). A megközelítések az anémia típusától és a hepcidinszint csökkentésének vagy növelésének céljától, illetve azok hatásától függően különböznek (2. táblázat).
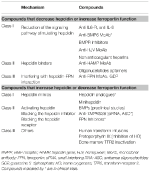
Táblázat 2. táblázat. A hepcidin-ferroportin tengelyt célzó kísérleti terápiák.
Kísérleti terápiák a hepcidinszint csökkentésére/a ferroportin funkció növelésére
A hepcidintermelő daganatok kivételével, amelyeket műtéti úton kell eltávolítani, a hepcidinszintet vagy annak hatásait antagonizáló vegyületek hasznosak lehetnek minden olyan anémiában, amelyet magas hepcidinszint jellemez. Fő alkalmazásuk a krónikus gyulladásos betegségekben lenne a hypoferrémiát és a vérszegénységet visszafordítani. Preklinikai vizsgálatokban számos kísérleti terápiát vizsgáltak, amelyek célja a hepcidin-útvonal és működésének manipulálása. A hepcidin-antagonisták a hepcidinszintézis gátlói/szabályozói (Ganz, 2019), a hepcidin működését gátló hepcidin-kötők és a hepcidin-ferroportin kölcsönhatást zavaró vegyületek (2. táblázat). Egyes vegyületek klinikai vizsgálatokban vannak, különösen krónikus vesebetegségben (Sheetz és mtsai., 2019). IRIDA-ban a hepcidin-útvonal manipulálását javasolták preklinikai vizsgálatokban anti-HJV MoAb alkalmazásával (Kovac és mtsai., 2016).
Kísérleti terápiák a hepcidinszint növelésére / a ferroportin funkció csökkentésére
A hepcidinszint növelése nemcsak a vastúlterhelést csökkentheti, hanem részben a vasterheléses anémiákban az ineffektív eritropoézist is kontrollálhatja. A β-talasszémia a leginkább vizsgált állapot ezek közül (Casu és mtsai., 2018; Gupta és mtsai., 2018). A javasolt gyógyszerek a hepcidin-analógok (egyesek klinikai vizsgálatokban), hepcidin-modulátorok, különösen a TMPRSS6 gátlók, vagy a hepcidin-ferroportin kölcsönhatásba beavatkozó, a vastartalmat csökkentő vegyületek (2. táblázat).
Míg a hepcidinszintet növelő vegyületek a nem hatékony erythropoiesis és a vasterhelés közötti ördögi kör miatt csökkentik az ineffektív erythropoiesist (Camaschella és Nai, 2016), az erythroid prekurzorok érését elősegítő gyógyszerek, mint az aktivinreceptor IIB ligandcsapda, a luspatercept, nemcsak az anaemiát javítják, hanem a hepcidin gátlásának csökkentésével a vas homeosztázist is javítják (Piga és mtsai, 2019).
A jelenleg klinikai vizsgálatokban lévő néhány célzott megközelítés remélhetőleg a különböző anémiák újszerű kezelését fogja eredményezni.
Következtetés
A vasanyagcsere és a hepcidin szabályozásának megértésében elért látványos előrelépések lehetővé tették az eritropoézis szabályozásának jobb megértését, mivel az eritropoetinnel együtt a vas az eritroid sejtek érésének alapvető tényezője. A vérszegénységhez vezető állapotok magas és alacsony hepcidinszintekhez társulhatnak. Mindkét esetben a hepcidin deregulációjának kontrasztos szabályozása javíthatja/javíthatja a vérszegénységet preklinikai modellekben, új eszközöket kínálva, amelyeket már most vagy hamarosan klinikailag is vizsgálnak a specifikus anémiák kezelésére.
Author Contributions
AP fogalmazta meg a cikket. CC dolgozta ki a végleges változatot. AN és LS hozzájárult a kézirat megírásához és kritikai felülvizsgálatához. Minden szerző jóváhagyta a végleges változatot.
Finanszírozás
Ezt a tanulmányt részben az EHA 2018-ban AP-nek nyújtott fejlett kutatási ösztöndíja támogatta. Cariplo Foundation Young Investigator Grant n° 2017-0916 to AN.
Conflict of Interest
CC a Vifor Pharma, a Celgene és a Novartis tanácsadója.
A többi szerző kijelenti, hogy a kutatást olyan kereskedelmi vagy pénzügyi kapcsolatok hiányában végezték, amelyek potenciális összeférhetetlenségként értelmezhetők.
An, P., Wu, Q., Wang, H., Guan, Y., Mu, M., Liao, Y., et al. (2012). A TMPRSS6, de nem a TF, TFR2 vagy BMP2 variánsok a vashiányos vérszegénység fokozott kockázatával járnak együtt. Hum. Mol. Genet. 21, 2124-2131. doi: 10.1093/hmg/dds028
PubMed Abstract | CrossRef Full Text | Google Scholar
Andriopoulos, B. Jr., Corradini, E., Xia, Y., Faasse, S. A., Chen, S., Grgurevic, L., et al. (2009). A BMP6 a hepcidin expresszió és a vasanyagcsere kulcsfontosságú endogén szabályozója. Nat. Genet. 41, 482-487. doi: 10.1038/ng.335
PubMed Abstract | CrossRef Full Text | Google Scholar
Arezes, J., Foy, N., McHugh, K., Sawant, A., Quinkert, D., Terraube, V., et al. (2018). Az eritroferron gátolja a hepcidin BMP6 általi indukcióját. Blood 132, 1473-1477. doi: 10.1182/blood-2018-06-857995
PubMed Abstract | CrossRef Full Text | Google Scholar
Aschemeyer, S., Qiao, B., Stefanova, D., Valore, E. V., Sek, A. C., Ruwe, T. A., et al. (2018). A ferroportin szerkezet-funkció analízise meghatározza a hepcidin kötőhelyét és alternatív hatásmechanizmusát. Blood. 131, 899-910. doi: 10.1182/blood-2017-05-786590
CrossRef Full Text | Google Scholar
Babitt, J. L., Huang, F. W., Wrighting, D. M., Xia, Y., Sidis, Y., Samad, T. A., et al. (2006). A hemojuvelin általi csontmorfogenetikus fehérje jelátvitel szabályozza a hepcidin expressziót. Nat. Genet. 38, 531-539. doi: 10.1038/ng1777
PubMed Abstract | CrossRef Full Text | Google Scholar
Beguin, Y. (2003). Oldható transzferrin receptor az eritropoézis és a vasstátusz értékelésére. Clin. Chim. Acta 329, 9-22. doi: 10.1016/S0009-8981(03)00005-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Bondu, S., Alary, A. S., Lefevre, C., Houy, A., Jung, G., Lefebvre, T., et al. (2019). Az eritroferron variáns megzavarja a vas homeosztázist SF3B1-mutációval járó myelodysplasztikus szindrómában. Sci. Transl. Med. 11:pii:eaav5467. doi: 10.1126/scitranslmed.aav5467
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C. (2013). Hogyan kezelem az atipikus mikrocitikus anémiában szenvedő betegeket. Br. J. Haematol. 160, 12-24. doi: 10.1111/bjh.12081
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C. (2019). Vashiány. Blood 133, 30-39. doi: 10.1182/blood-2018-05-815944
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C., and Nai, A. (2016). Hatástalan erythropoiesis és a vasstátusz szabályozása vasterheléses anaemiákban. Br. J. Haematol. 172, 512-523. doi: 10.1111/bjh.13820
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C., and Pagani, A. (2018). Előrelépések a vasanyagcsere és az eritropoézissel való kereszteződésének megértésében. Br. J. Haematol. 182, 481-494. doi: 10.1111/bjh.15403
PubMed Abstract | CrossRef Full Text | Google Scholar
Canali, S., Wang, C. Y., Zumbrennen-Bullough, K. B., Bayer, A., and Babitt, J. L. (2017). A Bone morphogenetic protein 2 a Bmp6-tól függetlenül szabályozza a vas homeosztázist egerekben. Am. J. Hematol. 92, 1204-1213. doi: 10.1002/ajh.24888
PubMed Abstract | CrossRef Full Text | Google Scholar
Casu, C., Nemeth, E., and Rivella, S. (2018). Hepcidin agonisták mint terápiás eszközök. Blood 131, 1790-1794. doi: 10.1182/blood-2017-11-737411
PubMed Abstract | CrossRef Full Text | Google Scholar
Colucci, S., Pagani, A., Pettinato, M., Artuso, I., Nai, A., Camaschella, C., et al. (2017). Az immunfilin FKBP12 gátolja a hepcidin expresszióját a BMP I-es típusú ALK2 receptor kötődésével hepatocitákban. Blood 130, 2111-2120. doi: 10.1182/blood-2017-04-780692
PubMed Abstract | CrossRef Full Text | Google Scholar
Crielaard, B. J., Lammers, T., and Rivella, S. (2017). A vasanyagcsere megcélzása a gyógyszerkutatásban és -szállításban. Nat. Rev. Drug Discov. 16, 400-423. doi: 10.1038/nrd.2016.248
PubMed Abstract | CrossRef Full Text | Google Scholar
De Falco, L., Sanchez, M., Silvestri, L., Kannengiesser, C., Muckenthaler, M. U., Iolascon, A., et al. (2013). Vasrefrakteres vashiányos vérszegénység. Haematologica 98, 845-853. doi: 10.3324/haematol.2012.075515
PubMed Abstract | CrossRef Full Text | Google Scholar
De Falco, L., Silvestri, L., Kannengiesser, C., Moran, E., Oudin, C., Rausa, M., et al. (2014). Új TMPRSS6 variánsok funkcionális és klinikai hatása vas-refrakter vashiányos vérszegénységben szenvedő betegeknél és genotípus-fenotípus vizsgálatok. Hum. Mutat. 35, 1321-1329. doi: 10.1002/humu.22632
PubMed Abstract | CrossRef Full Text | Google Scholar
Donker, A. E., Schaap, C. C., Novotny, V. M., Smeets, R., Peters, T. M., van den Heuvel, B. L., et al. (2016). Vasrefrakteres vashiányos vérszegénység: heterogén betegség, amely nem mindig vasrefrakteres. Am. J. Hematol. 91, E482-E490. doi: 10.1002/ajh.24561
PubMed Abstract | CrossRef Full Text | Google Scholar
Du, X., She, E., Gelbart, T., Truksa, J., Lee, P., Xia, Y., et al. (2008). A TMPRSS6 szerin proteáz szükséges a vashiány érzékeléséhez. Science 320, 1088-1092. doi: 10.1126/science.1157121
PubMed Abstract | CrossRef Full Text | Google Scholar
Finberg, K. E., Heeney, M. M., Campagna, D. R., Aydinok, Y., Pearson, H. A., Hartman, K. R., et al. (2008). A TMPRSS6 mutációi vas-refrakter vashiányos anémiát (IRIDA) okoznak. Nat. Genet. 40, 569-571. doi: 10.1038/ng.130
PubMed Abstract | CrossRef Full Text | Google Scholar
Ganz, T. (2019). A gyulladásos vérszegénység. N. Engl. J. Med. 381, 1148-1157. doi: 10.1056/NEJMra1804281
PubMed Abstract | CrossRef Full Text | Google Scholar
Gupta, R., Musallam, K. M., Taher, A. T., and Rivella, S. (2018). Hatástalan erythropoiesis: vérszegénység és vastúlterhelés. Hematol. Oncol. Clin. North Am. 32, 213-221. doi: 10.1016/j.hoc.2017.11.009
PubMed Abstract | CrossRef Full Text | Google Scholar
Heeney, M. M., and Finberg, K. E. (2014). Vas-refrakter vashiányos vérszegénység (IRIDA). Hematol. Oncol. Clin. North Am. 28, 637-652. doi: 10.1016/j.hoc.2014.04.009
PubMed Abstract | CrossRef Full Text | Google Scholar
Heeney, M. M., Guo, D., De Falco, L., Campagna, D. R., Olbina, G., Kao, P. P., et al. (2018). A hepcidin normalizálása előrejelzi a TMPRSS6 mutációs státuszt krónikus vashiányos betegeknél. Blood 132, 448-452. doi: 10.1182/blood-2017-03-773028
PubMed Abstract | CrossRef Full Text | Google Scholar
Kautz, L., Jung, G., Valore, E. V., Rivella, S., Nemeth, E., and Ganz, T. (2014). Az eritroferron mint a vasanyagcsere eritroid szabályozójának azonosítása. Nat. Genet. 46, 678-684. doi: 10.1038/ng.2996
PubMed Abstract | CrossRef Full Text | Google Scholar
Koch, P. S., Olsavszky, V., Ulbrich, F., Sticht, C., Demory, A., Leibing, T., et al. (2017). Az angiokrin Bmp2 jelátvitel egérmájban szabályozza a normális vas homeosztázist. Blood 129, 415-419. doi: 10.1182/blood-2016-07-729822
PubMed Abstract | CrossRef Full Text | Google Scholar
Kovac, S., Boser, P., Cui, Y., Ferring-Appel, D., Casarrubea, D., Huang, L., et al. (2016). Az anti-hemojuvelin antitest korrigálja a nem megfelelően magas hepcidinszint okozta vérszegénységet. Haematologica 101, e173-e176. doi: 10.3324/haematol.2015.140772
PubMed Abstract | CrossRef Full Text | Google Scholar
Lakhal, S., Schodel, J., Townsend, A. R., Pugh, C. W., Ratcliffe, P. J., and Mole, D. R. (2011). A II. típusú transzmembrán szerin proteináz TMPRSS6 hipoxia-indukálható faktorok általi szabályozása: új kapcsolat a hipoxia jelátvitel és a vas homeosztázis között. J. Biol. Chem. 286, 4090-4097. doi: 10.1074/jbc.M110.173096
PubMed Abstract | CrossRef Full Text | Google Scholar
Meynard, D., Kautz, L., Darnaud, V., Canonne-Hergaux, F., Coppin, H., and Roth, M. P. (2009). A BMP6 csontmorfogenetikus fehérje hiánya masszív vastúlterhelést indukál. Nat. Genet. 41, 478-481. doi: 10.1038/ng.320
PubMed Abstract | CrossRef Full Text | Google Scholar
Muckenthaler, M. U., Rivella, S., Hentze, M. W., and Galy, B. (2017). Vörös szőnyeg a vasanyagcserének. Cell 168, 344-361. doi: 10.1016/j.cell.2016.12.034
PubMed Abstract | CrossRef Full Text | Google Scholar
Nemeth, E., Tuttle, M. S., Powelson, J., Vaughn, M. B., Donovan, A., Ward, D. M., et al. (2004). A hepcidin a ferroportinhoz való kötődéssel és annak internalizációjának indukálásával szabályozza a celluláris vaskiáramlást. Science 306, 2090-2093. doi: 10.1126/science.1104742
PubMed Abstract | CrossRef Full Text | Google Scholar
Pagani, A., Colucci, S., Bocciardi, R., Bertamino, M., Dufour, C., Ravazzolo, R., et al. (2017). Az IRIDA új formája a TMPRSS6 és a BMP-receptort kódoló ACVR1A ALK2 kombinált heterozigóta mutációi miatt. Blood 129, 3392-3395. doi: 10.1182/blood-2017-03-773481
PubMed Abstract | CrossRef Full Text | Google Scholar
Pagani, A., Vieillevoye, M., Nai, A., Rausa, M., Ladli, M., Lacombe, C., et al. (2015). A sejtfelszíni transzferrin receptor-2 szabályozása vasfüggő hasítással és egy oldható forma felszabadításával. Haematologica 100, 458-465. doi: 10.3324/haematol.2014.118521
PubMed Abstract | CrossRef Full Text | Google Scholar
Pasricha, S. R., Lim, P. J., Duarte, T. L., Casu, C., Oosterhuis, D., Mleczko-Sanecka, K., et al. (2017). A hepcidin a promóter-asszociált hiszton acetiláció és a HDAC3 által szabályozott. Nat. Commun. 8:403. doi: 10.1038/s41467-017-00500-z
PubMed Abstract | CrossRef Full Text | Google Scholar
Piga, A., Perrotta, S., Gamberini, M. R., Voskaridou, E., Melpignano, A., Filosa, A., et al. (2019). A luspatercept javítja a hemoglobinszintet és a vérátömlesztési igényt egy béta-tahassziás betegek körében végzett vizsgálatban. Blood 133, 1279-1289. doi: 10.1182/blood-2018-10-879247
PubMed Abstract | CrossRef Full Text | Google Scholar
Rausa, M., Pagani, A., Nai, A., Campanella, A., Gilberti, M. E., Apostoli, P., et al. (2015). A Bmp6 expressziója egérmáj nem parenchimális sejtjeiben: a magas vaskivitelező aktivitásuk szabályozásának és a hepatociták vastúlterheléstől való védelmének mechanizmusa? PLoS One 10:e0122696. doi: 10.1371/journal.pone.0122696
PubMed Abstract | CrossRef Full Text | Google Scholar
Sebastiani, G., Wilkinson, N., and Pantopoulos, K. (2016). A hepcidin/ferroportin tengely farmakológiai célzása. Front. Pharmacol. 7:160. doi: 10.3389/fphar.2016.00160
PubMed Abstract | CrossRef Full Text | Google Scholar
Sheetz, M., Barrington, P., Callies, S., Berg, P. H., McColm, J., Marbury, T., et al. (2019). A hepcidin-ferroportin útvonal célzott kezelése a krónikus vesebetegség anaemiájában. Br. J. Clin. Pharmacol. 85, 935-948. doi: 10.1111/bcp.13877
PubMed Abstract | CrossRef Full Text | Google Scholar
Shore, E. M., Xu, M., Feldman, G. J., Fenstermacher, D. A., Cho, T. J., Choi, I. H., et al. (2006). A BMP I. típusú ACVR1 receptor visszatérő mutációja örökletes és sporadikus fibrodysplasia ossificans progressiva-t okoz. Nat. Genet. 38, 525-527. doi: 10.1038/ng1783
PubMed Abstract | CrossRef Full Text | Google Scholar
Silvestri, L., Pagani, A., Nai, A., De Domenico, I., Kaplan, J., and Camaschella, C. (2008). A szerin proteáz matriptáz-2 (TMPRSS6) gátolja a hepcidin aktiválódását a membrán hemojuvelin hasításával. Cell Metab. 8, 502-511. doi: 10.1016/j.cmet.2008.09.012
PubMed Abstract | CrossRef Full Text | Google Scholar
Silvestri, L., Rausa, M., Pagani, A., Nai, A., and Camaschella, C. (2013). Hogyan értékeljük a TMPRSS6 mutációk okozati összefüggéseit? Hum. Mutat. 34, 1043-1045. doi: 10.1002/humu.22321
PubMed Abstract | CrossRef Full Text | Google Scholar
Sonnweber, T., Nachbaur, D., Schroll, A., Nairz, M., Seifert, M., Demetz, E., et al. (2014). A hepcidin hipoxia indukálta downregulációját a vérlemezkékből származó BB növekedési faktor közvetíti. Gut 63, 1951-1959. doi: 10.1136/gutjnl-2013-305317
PubMed Abstract | CrossRef Full Text | Google Scholar
Sorensen, E., Rigas, A. S., Didriksen, M., Burgdorf, K. S., Thorner, L. W., Pedersen, O. B., et al. (2019). A hemoglobinszintet befolyásoló genetikai tényezők 15 567 véradóban: a dán véradói vizsgálat eredményei. Transfusion 59, 226-231. doi: 10.1111/trf.15075
CrossRef Full Text | Google Scholar
Theurl, I., Schroll, A., Sonnweber, T., Nairz, M., Theurl, M., Willenbacher, W., et al. (2011). A hepcidin expresszió farmakológiai gátlása visszafordítja a krónikus gyulladás okozta vérszegénységet patkányokban. Blood 118, 4977-4984. doi: 10.1182/blood-2011-03-345066
PubMed Abstract | CrossRef Full Text | Google Scholar
Weinstein, D. A., Roy, C. N., Fleming, M. D., Loda, M. F., Wolfsdorf, J. I., and Andrews, N. C. (2002). A hepcidin nem megfelelő expressziója vasrefrakteres anaemiához társul: következmények a krónikus betegségek anaemiájára. Blood 100, 3776-3781. doi: 10.1182/blood-2002-04-1260
PubMed Abstract | CrossRef Full Text | Google Scholar
Weiss, G., Ganz, T., and Goodnough, L. T. (2019). A gyulladásos vérszegénység. Blood 133, 40-50. doi: 10.1182/blood-2018-06-856500
PubMed Abstract | CrossRef Full Text | Google Scholar
Weiss, G., and Goodnough, L. T. (2005). A krónikus betegségek vérszegénysége. N. Engl. J. Med. 352, 1011-1023. doi: 10.1056/NEJMra041809
PubMed Abstract | CrossRef Full Text | Google Scholar
Leave a Reply