Frontiers in Physiology
Introduction
貧血は世界で最も多い疾患の1つで、複数の分析によると鉄欠乏による貧血が一般的です(Camaschella, 2019にレビューあり)。 このタイプの貧血は、全身の鉄不足と、組織の酸素化を維持するために十分な数の赤血球を産生するために骨髄が消費する大量の鉄を供給できないことから生じる
鉄利用率は、肝臓ペプチドホルモンのヘプシジンによって制御されている。 体内鉄の増加によりヘプシジンが産生され、循環中に放出され、腸球、マクロファージ、肝細胞に高発現する膜貫通型鉄輸出蛋白である受容体フェロポルティンに作用する。 ヘプシジンは、鉄の輸出を阻害し(Aschemeyerら、2018)、鉄輸出体フェロポーチンを分解することで吸収性十二指腸細胞や鉄リサイクリングマクロファージから血漿への鉄の侵入を抑制します(Nemethら、2004)。 血漿鉄および全身性鉄ホメオスタシスを調節することにより、ヘプシジン/フェロポーチン軸は赤血球生成に強く影響し、それゆえ貧血の発症の可能性がある<1531><3457>The Iron-Erythropoiesis Connection<9143><7863>赤血球生成プロセスは成熟赤血球のヘモグロビン合成に循環鉄量の約80%を消費している。 鉄の大部分(1日20~25mg)はマクロファージによって再利用され、限られた量(1日1~2mg)は腸管からの吸収に由来するものである。 腎臓ホルモンであるエリスロポエチン(EPO)は、赤血球前駆細胞の増殖、特にCFU-eと、より低い程度ではBFU-eの増殖、および末期赤血球形成の初期段階を制御する一方で、ヘモグロビンに組み込まれるヘムの合成のために、前赤芽球から網球までの分化後期の段階で鉄の必要量が増加する(Muckenthaler et al, 2017)。
ヘプシジン調節には、BMP-SMAD経路を活性化する骨形成タンパク質(BMP)を産生する肝内皮洞細胞(LSEC)とヘプシジンを産生・放出する肝細胞とのクロストークが必要である(Babitt et al.) BMP6とBMP2はヘプシジンをアップレギュレートする最も重要なBMPであるが、BMP6の発現は鉄依存性である(Andriopoulos et al., 2009; Meynard et al., 2009) BMP2は鉄反応性が低いようだ(Canali et al., 2017; Koch et al., 2017)<1531><7863>絶対鉄欠乏症および鉄欠乏症貧血ではヘプシジンのレベルが低くなっている。 これらの条件下では、鉄貯蔵量が枯渇し、BMP-SMADシグナル伝達が複数のレベルでスイッチオフされる。 まず、BMP6の発現が抑制され、次に、BMP共受容体ヘモジュベリンを切断するプロテアーゼであるTMPRSS6(Silvestri et al., 2008)の活性が強く上昇し(Lakhal et al., 2011)、3番目に、ヒストン脱アセチル化酵素3(HDAC3)がヘプシジン遺伝子座を抑制しています(Pasricha et al., 2017)。 鉄欠乏の状態では、ヘプシジン産生の減少は、食事および薬理学的な鉄吸収を促進する適応機構である(Camaschella and Pagani, 2018)。
貧血が深刻な場合、併存する低酸素は腎合成およびEPOの放出の増加を通じて赤血球造成を刺激する。 これは、赤芽球によって産生されるEPO標的遺伝子であるエリスロフェロン(ERFE)(Kautz et al., 2014)、他の組織によって放出される分子(例えば、PDGF-BB)(Sonnweber et al., 2014)、およびおそらくトランスフェリン受容体(TFR)、sTFR1(Beguin、2003)およびsTFR2(Pagani et al., 2015)の可溶成分によってヘプシジン転写を抑制させるに至った。 最終的な目的は、拡大した赤血球造血の必要性に十分な鉄を供給することである。
Anemias With Abnormal Hepcidin Levels
ヘプシジン値に基づいて、貧血はヘプシジンが高い貧血と低い貧血として分類されることがある。 ヘプシジン濃度が高い状態が続くと、鉄の吸収が阻害され、赤血球造血への鉄の供給が減少するため、鉄欠乏性貧血を引き起こすことは直感的に理解できる。 逆に、ヘプシジンが低値で鉄過剰の場合は、赤血球造血がうまくいかず、いわゆる鉄負荷性貧血を特徴とする。 これら2つの貧血は、相反する病態生理メカニズムの結果である(図1)。 第1群では、ヘプシジンが鉄の吸収と再利用に及ぼす抑制作用により、全身的な鉄欠乏が起こるため、第2群では、拡大した異常赤血球生成によりヘプシジンが抑制されるために貧血となる(Camaschella and Nai, 2016)
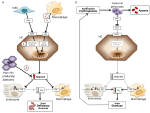
図1. ヘプシジンが高い貧血(左図)と低い貧血(右図)のメカニズムを模式的に示す。 パネル(A)。 ヘプシジン高値に伴う貧血の分子的な病態。 LSEC、骨形成タンパク質(BMP)を産生する肝類洞内皮細胞、BMPRs、BMP受容体、IL6、インターロイキン6、HC、肝細胞、HAMP、ヘプシジン遺伝子。 Fe、鉄;FPN、フェロポーチン;1、IRIDA;2、炎症性貧血;3、ヘプシジン産生腺腫。 パネル(B)。 赤血球生成不全による貧血におけるヘプシジン変異の分子的病態。 ERFE、エリスロフェロン封鎖型BMP。 トランスフェリン飽和度の低下や低酸素症など、この種の貧血でヘプシジンを阻害する他の機序は示していない。 詳細は本文を参照。
Anemia Associated With High Hepcidin Levels
このグループには2つの遺伝性希少疾患(鉄不応性鉄欠乏性貧血と先天的な糖代謝異常のヘプシジン生成腺腫)および後天的によくみられる疾患:炎症性の貧血が含まれる(表1)。 ヘプシジン値によって分類される貧血症。
鉄難治性鉄欠乏性貧血
鉄難治性鉄欠乏性貧血(IRIDA)は低色素性小球性貧血、低いトランスフェリン飽和、不適切に正常/高いヘプシジン値で特徴づけられるまれな劣性疾患である。 この疾患は、II型セリンプロテアーゼであるマトリプターゼ-2をコードする遺伝子TMPRSS6 (Finberg et al., 2008) の変異によって引き起こされます (Du et al., 2008)。 TMPRSS6の変異は遺伝子に沿って広がっており、異なるドメイン、特に触媒ドメインに影響を及ぼす可能性があります(De Falco et al.) この膜貫通型プロテアーゼは肝臓で高発現しており、細胞表面のBMP共受容体ヘモジュベリンを切断することでヘプシジンの転写を阻害し、BMPシグナルとヘプシジン合成を減衰させる(Silvestri et al.、2008年)。 TMPRSS6の機能は、鉄の吸収を増加させる代償機構を可能にするために、鉄欠乏症では必須です。
イリダは出生時から存在し、通常は小児期に診断されます。 古典的な鉄欠乏症と比較して、鉄のパラメータが非典型的であるため、本疾患を疑うことになる。 トランスフェリン飽和率は他の鉄欠乏症と同様に強く低下(10%未満)しますが、鉄欠乏症とは異なり、血清フェリチンのレベルは正常または上昇します(Camaschella, 2013; De Falco et al.)
イリーダ診断のために提案された検査はいずれも100%の症例をカバーしていない。 遺伝子検査では、TMPRSS6変異が特定されるが、その中には(ノンセンス変異、フレームシフト変異、スプライシング変異)、明らかに原因となっているものがある。 他のケースでは、これまで報告されていないミスセンス変異のように、因果関係を証明するために機能研究が必要である(Silvestri et al.、2013)。 しかし、これらの検査はほとんど行われていない。 血清ヘプシジン値は通常、鉄欠乏とは無関係に増加/正常であり、フェリチンの高値/正常値と一致する。 1531>
難治性鉄欠乏症の表現型を持つ患者の中には、TMPRSS6変異アレルを1つ持つ患者が報告されているが、ここではIRIDAとみなすべきかどうかが議論されている。 TMPRSS6ホモ接合体あるいは複合ヘテロ接合体変異による古典的な重症IRIDAから、単一の変異/多型変化による鉄欠乏症感受性の上昇まで、さまざまな病態が想定される。 古典的IRIDAを予測するために提案された1つのアプローチは、トランスフェリン飽和度(Tsat)/logヘプシジンまたはTsat/logフェリチンの比率として、他の鉄パラメータ上のヘプシジン正常化である(Donkerら、2016年)。 他の著者によると、重度のIRIDA表現型を持つ患者のほとんどは、二遺伝子性TMPRSS6変異を有し、未同定の場合、第2の対立遺伝子は遺伝的に潜んでいる可能性がある(Heeny et al.、2018年)。 一般論として、単一のアレルを有する対象は、2つの変異を有する対象よりも表現型が穏やかであり、鉄治療によく反応する(Donkerら、2016年)。 興味深いことに、いくつかのTMPRSS6 SNPは、いくつかの集団(An et al., 2012)および献血者(Sorensen et al., 2019)において鉄欠乏に対する感受性を与えることが示されている<1531><7863>もともとTMPRSS6(I212T)の原因変異1つと(R271Q)サイレント変異1つを有する非定型IRIDA遺伝子型であることがわかった5歳の女性において、二遺伝子継承が報告されている(De Falco et al, 2014)。 その後、ALK2をコードするBMP I型受容体遺伝子ACVR1の変異により、軟部組織に異所性骨形成を伴う稀な優性疾患であるFibrodysplasia ossificans progressiva(FOP)と診断された(Shore et al.、2006年)。 病的対立遺伝子ALK2R258Sは、変異が遺伝子のグリシンセリンリッチドメインに影響を与え、BMP/SMAD経路がその特異的阻害剤FKBP12と結合できない過活動状態になるため、構成的に活性である(Pagani et al.、2017)
この稀なケースは特に例証となるものだ。 まず、ALK2のグリシンセリンリッチドメインはFKBP12と相互作用し、この変異は結合を不安定にするので、肝臓ALK2およびヘプシジンの調節因子としてFKBP12のこれまで疑われていなかった役割を明らかにした(Colucci et al.、2017年)。 第二に、骨と肝臓のBMPタイプI受容体の活性化の関連性を明らかにすることにつながりました。 第三に、同じACVR1変異と推定される正常なTMPRSS6を有する他のFOP患者の間でIRIDAが確認されなかったため、本件は肝BMP/SMADシグナルの制御における無傷のTMPRSS6の関連性を強化した(Pagani et al.、2017年)。 最後に,本例はTMPRSS6ハプロイン不全が古典的なIRIDAを引き起こすことはできないという概念と一致している<1531><7863>IRIDAの最適な治療法は未定である。 鉄の経口投与は吸収されないため、効果がない。 ビタミンCの添加により、散発的な効果が期待できる。 鉄剤の静脈注射は、後天性鉄欠乏症の患者と比較して、通常、遅い速度で部分的な反応を引き起こす。 EPOは古典的な症例では効果がない(De Falcoら、2013;Heeny and Finberg、2014)。
ヘプシジン産生腺腫貧血
これはグリコーゲン分解とグルコゲン生成に関わる反応を触媒するグルコース6ホスファターの欠損による劣性障害、グリコーゲン貯蔵疾患1aに罹患する成人の極めて珍しい症状である。 低血糖が危険な症状としてよく知られています。 現在の治療では、患児は成人まで生存できるが、貧血や肝臓腺腫などの合併症が発生する。 貧血は微小球性低色素性で、鉄欠乏性であり、鉄の経口投与に抵抗性である。 貧血は外科的な腺腫の切除後に回復した。 腺腫の組織ではヘプシジン mRNA が陽性であったが、正常な周辺組織ではヘプシジンの抑制が認められ、異所性の制御不能なヘプシジン産生のためと考えられた(Weinstein ら、2002)。 患者の血液学的特徴は、貧血の共通の機序として高ヘプシジン値を示すことからIRIDAと類似している。
炎症性貧血
従来慢性疾患の貧血として知られていた炎症性貧血(AI)は、全身性の炎症と免疫活性化の状態で発症する中程度の正常色調-正常細胞性貧血である。 慢性感染症、自己免疫疾患、進行性癌、慢性腎臓病、うっ血性心不全、慢性閉塞性肺疾患、高齢者の貧血(少なくとも一部)、移植片対宿主病など、いくつかの共通の疾患において発症する。 AIは世界で最も一般的な貧血の一つであり、入院患者における最も頻度の高い貧血でもあります。 急性炎症は、集中治療室における貧血の重症化に寄与しています。 AIの基礎となる分子メカニズムは複数あり、複雑である。 マクロファージによるIL1-β、TNF-α、IL-6などのサイトカインやリンパ球によるINF-γの過剰産生はEPO産生を鈍らせ、赤血球生成反応を損ない、ヘプシジンレベルを上昇させ、特に急性型では赤血球貪食を活性化することがあります(Weiss and Goodnough, 2005; Ganz, 2019)。
ヘプシジンは、IL-6受容体(IL-6R)およびJAK2-STAT3シグナルを介して、IL-6によって活性化される。 BMPシグナルの不活性化は、炎症の動物モデルにおいてヘプシジンを減少させるため、完全なヘプシジン活性化には活発なBMP-SMAD経路が必要である(Theurlら、2011年)。 全身の鉄のホメオスタシスのデレギュレーションは、マクロファージの鉄の隔離と吸収・リサイクルの減少を引き起こし、トランスフェリンの低飽和と赤血球生成や他の組織の鉄制限につながる。
AIの伝統的な治療は、可能な限り基礎疾患の回復/制御に基づいて行われる。 疾患が治療不可能で貧血が軽度であれば、どのような治療でも副作用を避けるために、リスクとベネフィットを慎重に評価する必要がある。 病態生理に基づく治療は、エリスロポエチン様化合物と鉄に限定される。 赤血球生成促進剤(ESA)の使用は、赤血球の拡大を誘導することによりヘプシジンを抑制する。 この方法は、慢性腎臓病、低リスクの骨髄異形成症候群、化学療法中の癌患者などに広く用いられている。 しかし、高用量には心血管系の副作用があるため、慎重な臨床管理が必要である。 鉄の静脈内投与は、ESA依存性の赤血球増加による鉄制限を緩和する可能性がある。 鉄の経口投与は、ヘプシジン濃度が高いために腸管吸収が阻害されるため、通常、効果がない。 プロリル水酸化酵素の阻害剤(低酸素誘導因子、HIF安定化剤)は、内因性EPOを増加させる目的で、慢性腎臓病で実験的に使用されています。 赤血球輸血による慢性的な治療は、一過性の効果と副作用のために推奨されておらず、重度の難治性貧血に限定されている(Camaschella, 2019; Weiss et al., 2019)。
低ヘプシジンレベルに関連した貧血
有効でない赤血球生成と低ヘプシジンまたは不適切に正常で、結果として鉄過多になることは「鉄負荷性貧血」の特徴である。” 原型はβ-サラセミアで、β-グロビン遺伝子の変異により、貧血とα-グロビン鎖の過剰産生を引き起こす遺伝的劣性疾患である。 βグロビン鎖は骨髄中にヘミクロムとして析出し、成熟した赤血球前駆体を損傷し、赤血球造血能が低下する。 これは、非輸血依存性サラセミアまたはサラセミア中間体において起こり、その赤血球生成は、肝臓のヘプシジン発現を抑制するためにエリスロフェロンを放出する未熟細胞の優勢によって特徴付けられる。 ヘプシジンレベルは通常、輸血依存性サラセミアでより大きく、内因性非有効赤血球造血が輸血によって少なくとも部分的に抑制される(Camaschella and Nai, 2016)。
ヘプシジン抑制は、EPO刺激により赤芽球によって合成されるERFE遺伝子によりコード化されるTNF-αファミリーのメンバーのサイトカインエリスロフェロン(ERFE)増加が媒介となる(Kautz et al.、2014)。 ERFEは循環中に放出され、BMP、特にBMP6を封じ込め(Arezes et al., 2018)、鉄に反応するヘプシジンシグナルを減衰させる。 さらに、ヒストン脱アセチル化酵素HDAC3によってヘプシジン遺伝子座にエピジェネティックな抑制が起こる(Pasricha et al.、2017)。 貧血が低酸素を引き起こすと、PDGF-BBなどの他のメディエーター(Sonnweber et al, 2014)、異なる細胞タイプによって放出される、ヘプシジンを抑制する。
スプライソソーム遺伝子SF3B1の変異によるクローン障害である環状鉄芽球を伴う低リスクの骨髄異形成において、ヘプシジンレベルは特殊なメカニズムで減少する。 ミトコンドリアに鉄が蓄積して非効率な赤血球形成と全身の鉄過剰に至る。 異常にスプライスされた伸長型ERFEタンパク質は、野生型ERFEよりも強力にヘプシジンを抑制する(Bondu et al, 2019)、輸血非依存性鉄負荷を引き起こす。
Targeted Therapies for Hepcidin-Related Anemias
先に述べた貧血を引き起こす分子メカニズムの特定は、現在の対症療法を代替する標的療法の開発の研究を刺激した(セバスチャーニら、2016;Crielaardら、2017)。 貧血の種類やヘプシジンレベルの減少や増加、またはその効果を目的とするものによってアプローチが異なる(表2)
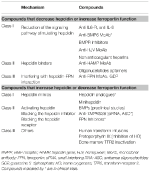
Table 2. ヘプシジン-フェロポーチン軸を標的とする実験的療法
ヘプシジンレベルを減少させる/フェロポーチン機能を増加させる実験的療法
外科的に除去しなければならないヘプシジン生成腫瘍を除いて、ヘプシジンまたはその効果を拮抗する化合物は高いヘプシジンレベルによって特徴付けられるすべての貧血に有用であると考えられる。 その主な用途は、低フェロモン血症と貧血を回復させるための慢性炎症性疾患であろう。 ヘプシジン経路とその機能を操作することを目的としたいくつかの実験的治療法が、前臨床試験で研究されている。 ヘプシジン拮抗薬は、ヘプシジン合成/調節因子の阻害剤(Ganz, 2019)、その機能を阻害するヘプシジン結合剤、およびヘプシジン-フェロポーティン相互作用を阻害する化合物である(表2)。 いくつかの化合物は、特に慢性腎臓病において臨床試験中である(Sheetz et al.、2019)。 IRIDAでは、抗HJV MoAbを用いた前臨床試験でヘプシジン経路の操作が提案されている(Kovacら、2016)
ヘプシジンレベルを増加/フェロポーチン機能を低下させる実験療法
ヘプシジンレベルを増加させると、鉄過剰負荷を軽減するだけではなく、鉄負荷貧血における非効率な赤血球生成も一部制御できると考えられる。 β-サラセミアは、これらの状態の中で最も研究されています(Casuら、2018;Guptaら、2018)。 提案されている薬剤は、ヘプシジンアナログ(一部は臨床試験中)、ヘプシジンモジュレーター、特にTMPRSS6阻害剤、または鉄輸出を減少させるヘプシジン-フェロポーティン相互作用を妨害する化合物である(表2)。
ヘプシジンを増加させる化合物は、非効率的な赤血球生成と鉄負荷の間の悪循環により非効率的な赤血球生成を減少させるが(Camaschella and Nai, 2016)、アクチビン受容体IIBリガンドトラップ、ルスパテルセプトのように、赤血球前駆体の成熟を促進する薬剤は、ヘプシジン抑制を減らすことにより、貧血を改善するだけではなく、鉄恒常性も向上させる(Piga et al, 2019)。
現在臨床試験中のいくつかの標的アプローチにより、さまざまな貧血に対する新規治療法が生まれることを期待する。
結論
鉄代謝およびヘプシジンの制御の理解における目覚ましい進歩は、エリスロポエチンと共に鉄は赤血球成熟の基本因子なので、赤血球生成制御についての理解を深めることができるようになった。 貧血を引き起こす疾患は、ヘプシジンレベルが高い場合と低い場合がある。 ヘプシジンの調節は、前臨床モデルにおいて貧血を改善・抑制する可能性があり、特定の貧血の治療のために、すでに臨床的に検討されている、あるいは近い将来検討されるであろう新しい手段を提供するものです。 最終版はCCが作成した。 ANとLSは原稿の執筆とクリティカルレビューに貢献した。 すべての著者が最終版を承認した。
Funding
本論文は、APに対する2018年のEHA Advanced Research Grantによって部分的に支援された。 Cariplo Foundation Young Investigator Grant n° 2017-0916 to AN.
Conflict of Interest
CC is a consultant of Vifor Pharma, Celgene, and Novartis.
The remaining authors declare that the research was conducted in absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
An, P…, Wu, Q., Wang, H., Guan, Y., Mu, M., Liao, Y.、他 (2012). TMPRSS6、しかしTF、TFR2またはBMP2の変異は鉄欠乏性貧血のリスク増加と関連している。 Hum. Mol. Genet. 21, 2124-2131. doi: 10.1093/hmg/dds028
PubMed Abstract | CrossRef Full Text | Google Scholar
Andriopoulos, B. Jr., Corradini, E., Xia, Y., Faasse, S. A., Chen, S., Grgurevic, L.ら(2009年). BMP6はヘプシジンの発現と鉄代謝の重要な内因性調節因子である。 Nat. Genet. 41, 482-487. doi: 10.1038/ng.335
PubMed Abstract | CrossRef Full Text | Google Scholar
Arezes, J., Foy, N., McHugh, K., Sawant, A., Quinkert, D., Terraube, V.ら (2018).Soc. エリスロフェロンは、BMP6によるヘプシジンの誘導を抑制する。 Blood 132, 1473-1477. doi: 10.1182/blood-2018-06-857995
PubMed Abstract | CrossRef Full Text | Google Scholar
Aschemeyer, S., Qiao, B., Stefanova, D., Valore, E.V., Sek, A. C., Ruwe, T. A., et al. (2018). フェロポルティンの構造機能解析により、ヘプシジンの結合部位と代替作用機序が明らかになった。 Blood. 131, 899-910.doi: 10.1182/blood-2017-05-786590
CrossRef Full Text | Google Scholar
Babitt, J. L., Huang, F. W. , Wrighting, D. M. , Xia, Y. , Sidis, Y. , Samad, T. A. , et al. ヘモジュベリンによる骨形成タンパク質シグナルは、ヘプシジンの発現を制御する。 Nat. Genet. 38, 531-539. doi: 10.1038/ng1777
PubMed Abstract | CrossRef Full Text | Google Scholar
Beguin, Y. (2003). 赤血球造血と鉄の状態を評価するための可溶性トランスフェリン受容体。 Clin. Chim. Acta 329, 9-22. doi: 10.1016/S0009-8981(03)00005-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Bondu, S., Alary, A. S., Lefevre, C., Houy, A., Jung, G., Lefebvre, T., et al (2019). バリアントエリスロフェロンは、SF3B1変異骨髄異形成症候群の鉄のホメオスタシスを破壊する。 Sci. Transl. Med. 11:pii:eaav5467. doi: 10.1126/scitranslmed.aav5467
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C. (2013). 非定型小球性貧血の患者をどう管理するか。 Br. J. Haematol. 160, 12-24. doi: 10.1111/bjh.12081
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella、C. (2019).(英語). 鉄欠乏症。 Blood 133, 30-39. doi: 10.1182/blood-2018-05-815944
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C., and Nai, A. (2016). 鉄負荷貧血症における非効率的な赤血球生成と鉄状態の調節。 Br. J. Haematol. 172, 512-523. doi: 10.1111/bjh.13820
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella,C., and Pagani,A. (2018). 鉄代謝とその赤血球造血とのクロストークを理解するための進歩。 Br. J. Haematol. 182, 481-494. doi: 10.1111/bjh.15403
PubMed Abstract | CrossRef Full Text | Google Scholar
Canali, S., Wang, C. Y., Zumbrennen-Bullough, K. B., Bayer, A. and Babitt, J. L.(2017).[日本版注釈] (2017). 骨形成タンパク質2はBmp6に依存しないマウスの鉄ホメオスタシスを制御する。 Am. J. Hematol. 92, 1204-1213. doi: 10.1002/ajh.24888
PubMed Abstract | CrossRef Full Text | Google Scholar
Casu, C., Nemeth, E., and Rivella, S. (2018). 治療ツールとしてのヘプシジンアゴニスト。 Blood 131, 1790-1794. doi: 10.1182/blood-2017-11-737411
PubMed Abstract | CrossRef Full Text | Google Scholar
Colucci,S., Pagani,A., Pettinato,M., Artuso,I., Nai,A., Camaschella,C., et al. (2017). イムノフィリンFKBP12は、肝細胞においてBMPタイプI受容体ALK2と結合することによりヘプシジンの発現を抑制する。 Blood 130, 2111-2120. doi: 10.1182/blood-2017-04-780692
PubMed Abstract | CrossRef Full Text | Google Scholar
Crielaard, B. J., Lammers, T., and Rivella, S. (2017). 創薬とデリバリーにおける鉄代謝の標的化。 Nat. Rev. Drug Discov. 16, 400-423. doi: 10.1038/nrd.2016.248
PubMed Abstract | CrossRef Full Text | Google Scholar
De Falco, L., Sanchez, M., Silvestri, L., Kannengiesser, C., Muckenthaler, M. U., Iolascon, A.ら (2013). 鉄剤不応性鉄欠乏性貧血。 Haematologica 98, 845-853. doi: 10.3324/haematol.2012.075515
PubMed Abstract | CrossRef Full Text | Google Scholar
De Falco, L., Silvestri, L., Kannengiesser, C., Moran, E., Oudin, C., Rausa, M.、その他 (2014).Kanngiesser (2014). 鉄難治性鉄欠乏性貧血患者における新規TMPRSS6バリアントの機能的および臨床的影響と遺伝子型-表現型研究。 Hum. Mutat. 35, 1321-1329. doi: 10.1002/humu.22632
PubMed Abstract | CrossRef Full Text | Google Scholar
Donker, A. E., Schaap, C. C., Novotny, V. M., Smeets, R., Peters, T. M., van den Heuvel, B. L., et al. (2016). 鉄不応性鉄欠乏性貧血:必ずしも鉄不応性とは限らない異質な疾患。 Am. J. Hematol. 91, E482-E490. doi: 10.1002/ajh.24561
PubMed Abstract | CrossRef Full Text | Google Scholar
Du, X., She, E., Gelbart, T., Truksa, J., Lee, P., Xia, Y.、他 (2008). セリンプロテアーゼTMPRSS6は、鉄欠乏を感知するのに必要である。 Science 320, 1088-1092. doi: 10.1126/science.1157121
PubMed Abstract | CrossRef Full Text | Google Scholar
Finberg, K. E., Heeney, M. M., Campagna, D. R., Aydinok, Y., Pearson, H. A., Hartman, K. R., and al. (2008). TMPRSS6の変異は、鉄不応性鉄欠乏性貧血(IRIDA)を引き起こす。 Nat. Genet. 40, 569-571. doi: 10.1038/ng.130
PubMed Abstract | CrossRef Full Text | Google Scholar
Ganz, T. (2019). 炎症の貧血 N. Engl. J. Med. 381, 1148-1157. doi: 10.1056/NEJMra1804281
PubMed Abstract | CrossRef Full Text | Google Scholar
Gupta, R., Musallam, K. M., Taher, A. T. , and Rivella, S. (2018). 非効率的な赤血球生成:貧血と鉄過多. Hematol. Oncol. Clin. North Am. 32, 213-221. doi: 10.1016/j.hoc.2017.11.009
PubMed Abstract | CrossRef Full Text | Google Scholar
Heeney, M. M., and Finberg, K.E. (2014). 鉄不応性鉄欠乏性貧血(IRIDA)。 Hematol. Oncol. Clin. North Am. 28, 637-652. doi: 10.1016/j.hoc.2014.04.009
PubMed Abstract | CrossRef Full Text | Google Scholar
Heeney, M.M., Guo, D.De Falco, L., Campagna, D. R., Olbina, G.、Kao, P. P., et al.(2018).の項参照。 ヘプシジンの正常化は、慢性鉄欠乏症患者におけるTMPRSS6変異の状態を予測する。 Blood 132, 448-452. doi: 10.1182/blood-2017-03-773028
PubMed Abstract | CrossRef Full Text | Google Scholar
Kautz, L., Jung, G., Valore, E. V., Rivella, S., Nemeth, E., and Ganz, T. (2014). 鉄代謝の赤血球制御因子としてのエリスロフェロンの同定。 Nat. Genet. 46, 678-684. doi: 10.1038/ng.2996
PubMed Abstract | CrossRef Full Text | Google Scholar
Koch, P. S., Olsavszky, V., Ulbrich, F., Sticht, C., Demory, A., Leibing, T, and al. (2017). マウス肝臓におけるアンジオクリンBmp2シグナルは、正常な鉄のホメオスタシスを制御している。 Blood 129, 415-419. doi: 10.1182/blood-2016-07-729822
PubMed Abstract | CrossRef Full Text | Google Scholar
Kovac,S., Boser,P., Cui,Y., Ferring-Appel,D., Casarrubea,D., Huang,L., et al(2016).鉄の正常な恒常性を制御する。 抗ヘモジュベリン抗体は不適切に高いヘプシジンレベルによって引き起こされる貧血を修正する。 Haematologica 101, e173-e176. doi: 10.3324/haematol.2015.140772
PubMed Abstract | CrossRef Full Text | Google Scholar
Lakhal, S., Schodel, J., Townsend, A. R., Pugh, C. W., Ratcliffe, P. J., and Mole, D. R. (2011). 低酸素誘導因子によるII型膜貫通型セリンプロテアーゼTMPRSS6の制御:低酸素シグナルと鉄ホメオスタシスとの間の新たな関連性。 J. Biol. Chem. 286, 4090-4097. doi: 10.1074/jbc.M110.173096
PubMed Abstract | CrossRef Full Text | Google Scholar
Meynard, D., Kautz, L., Darnaud, V., Canonne-Hergaux, F., Coppin, H. and Roth, M. P. (2009).の項参照。 骨形成タンパク質BMP6の欠失は、大量の鉄過剰症を誘発する。 Nat. Genet. 41, 478-481. doi: 10.1038/ng.320
PubMed Abstract | CrossRef Full Text | Google Scholar
Muckenthaler, M. U., Rivella, S., Hentze, M. W., and Galy, B.(2017).[日本語訳]骨形成タンパク質の欠失は、大規模な鉄過剰症を引き起こす。 鉄代謝のための赤い絨毯。 Cell 168, 344-361. doi: 10.1016/j.cell.2016.12.034
PubMed Abstract | CrossRef Full Text | Google Scholar
Nemeth, E., Tuttle, M. S., Powelson, J., Vaughn, M. B., Donovan, A., Ward, D.M., and al. (2004). ヘプシジンはフェロポルティンに結合し、その内在化を誘導することにより、細胞内の鉄排出を制御する。 サイエンス 306, 2090-2093.1104742
PubMed Abstract | CrossRef Full Text | Google Scholar
Pagani, A., Colucci, S., Bocciardi, R., Bertamino, M., Dufour, C., Ravazzolo, R., et al. (2017). BMP受容体ALK2をコードするTMPRSS6とACVR1Aの複合ヘテロ接合型変異による新しい型のIRIDA Blood 129, 3392-3395. doi: 10.1182/blood-2017-03-773481
PubMed Abstract | CrossRef Full Text | Google Scholar
Pagani, A…, Vieillevoye, M., Nai, A., Rausa, M., Ladli, M., Lacombe, C., et al. (2015). 鉄依存的な切断と可溶型の放出による細胞表面トランスフェリン受容体-2の制御。 Haematologica 100, 458-465. doi: 10.3324/haematol.2014.118521
PubMed Abstract | CrossRef Full Text | Google Scholar
Pasricha, S. R., Lim, P. J., Duarte, T. L., Casu, C., Oosterhuis, D., Mleczko-Sanecka, K.,など (2017). ヘプシジンはプロモーターに関連したヒストンアセチル化とHDAC3によって制御される。 Nat. Commun. 8:403. doi: 10.1038/s41467-017-00500-z
PubMed Abstract | CrossRef Full Text | Google Scholar
Piga, A., Perrotta, S., Gamberini, M. R., Voskaridou, E., Melpignano, A., Filosa, A., et al(2019).の項参照。 ルスパテルセプトは、βサラセミア患者を対象とした試験において、ヘモグロビン値と輸血の必要性を改善する。 Blood 133, 1279-1289. doi: 10.1182/blood-2018-10-879247
PubMed Abstract |Ref Full Text | Google Scholar
Rausa, M., Pagani, A., Nai, A., Campanella, A., Gilberti, M.E., Apostoli, P.、他 (2015). マウス肝非実質細胞におけるBmp6発現:その高い鉄輸出活性を制御し、鉄過剰負荷から肝細胞を保護するメカニズム? PLoS One 10:e0122696. doi: 10.1371/journal.pone.0122696
PubMed Abstract | CrossRef Full Text | Google Scholar
Sebastiani, G., Wilkinson, N., and Pantopoulos, K.(2016).鉄の過負荷からの保護。 ヘプシジン/フェロポーティン軸の薬理学的標的化。 Front. Pharmacol. 7:160. doi: 10.3389/fphar.2016.00160
PubMed Abstract | CrossRef Full Text | Google Scholar
Sheetz, M., Barrington, P., Callies, S., Berg, P. H., McColm, J., Marbury, T., et al(2019).。 慢性腎臓病の貧血におけるヘプシジン-フェロポーチン経路の標的化。 Br. J. Clin. Pharmacol. 85, 935-948. doi: 10.1111/bcp.13877
PubMed Abstract | CrossRef Full Text | Google Scholar
Shore, E. M., Xu, M., Feldman, G. J., Fenstermacher, D. A., Cho, T. J., Choi, I. H.ら(2006年)。 BMPタイプI受容体ACVR1の再発性変異は、遺伝性及び散発性の進行性骨化性線維異形成症を引き起こす。 Nat. Genet. 38, 525-527. doi: 10.1038/ng1783
PubMed Abstract | CrossRef Full Text | Google Scholar
Silvestri, L., Pagani, A., Nai, A., De Domenico, I. , Kaplan, J., and Camaschella, C. (2008). セリンプロテアーゼ・マトリプターゼ-2(TMPRSS6)は膜ヘモジュベリンを切断することでヘプシジンの活性化を阻害する。 セル・メタブ。 8, 502-511. doi: 10.1016/j.cmet.2008.09.012
PubMed Abstract | CrossRef Full Text | Google Scholar
Silvestri, L., Rausa, M., Pagani, A., Nai, A., and Camaschella, C. (2013). TMPRSS6変異の因果関係をどう評価するか? Hum. Mutat. 34, 1043-1045. doi: 10.1002/humu.22321
PubMed Abstract | CrossRef Full Text | Google Scholar
Sonnweber, T., Nachbaur, D., Schroll, A., Nairz, M., Seifert, M., Demetz, E., and al. (2014). 低酸素によるヘプシジンのダウンレギュレーションは、血小板由来成長因子BBによって媒介される。 Gut 63, 1951-1959. doi: 10.1136/gutjnl-2013-305317
PubMed Abstract | CrossRef Full Text | Google Scholar
Sorensen, E., Rigas, A. S., Didriksen, M., Burgdorf, K. S., Thorner, L. W., Pedersen, O. B., et al(2019).日本学術会議会員(会員番号:9000)。 15,567人の献血者におけるヘモグロビン値に影響を及ぼす遺伝的要因:デンマーク献血者調査の結果。 Transfusion 59, 226-231. doi: 10.1111/trf.15075
CrossRef Full Text | Google Scholar
Theurl, I., Schroll, A., Sonnweber, T., Nairz, M., Willenbacher, W.他 (2011). ヘプシジン発現の薬理学的阻害は、ラットの慢性炎症による貧血を回復させる。 Blood 118, 4977-4984. doi: 10.1182/blood-2011-03-345066
PubMed Abstract | CrossRef Full Text | Google Scholar
Weinstein, D. A., Roy, C. N., Fleming, M. D., Loda, M. F., Wolfsdorf, J. I., and Andrews, N. C. (2002). ヘプシジンの不適切な発現は鉄難病性貧血と関連している:慢性疾患の貧血への示唆。 Blood 100, 3776-3781. doi: 10.1182/blood-2002-04-1260
PubMed Abstract | CrossRef Full Text | Google Scholar
Weiss, G., Ganz, T., and Goodnough, L. T. (2019). 炎症の貧血。 Blood 133, 40-50. doi: 10.1182/blood-2018-06-856500
PubMed Abstract | CrossRef Full Text | Google Scholar
Weiss, G., and Goodnough, L. T. (2005).貧血。 慢性疾患の貧血。 N. Engl. J. Med. 352, 1011-1023. doi: 10.1056/NEJMra041809
PubMed Abstract | CrossRef Full Text | Google Scholar
.
Leave a Reply