Frontiers in Physiology
Introduction
Anemia jest jednym z najczęstszych zaburzeń na całym świecie, a niedokrwistość spowodowana niedoborem żelaza jest przeważającą formą według wielu analiz (przegląd w Camaschella, 2019). Ten rodzaj niedokrwistości wynika z całkowitego niedoboru żelaza w organizmie i niezdolności do dostarczenia dużej ilości żelaza, które szpik kostny zużywa do produkcji odpowiedniej liczby czerwonych krwinek w celu utrzymania natlenienia tkanek.
Dostępność żelaza jest kontrolowana przez wątrobowy hormon peptydowy hepcidin. Wzrost żelaza w organizmie powoduje produkcję hepcydyny, która jest uwalniana do krążenia i działa na jej receptor ferroportynę, transmembranowe białko eksportera żelaza wysoko wyrażone na enterocycie, makrofagach i hepatocytach. Hepcydyna zmniejsza przedostawanie się żelaza do osocza z absorpcyjnych komórek dwunastnicy i makrofagów recyklingu żelaza poprzez blokowanie eksportu żelaza (Aschemeyer i in., 2018) i poprzez degradację ferroportyny eksportera żelaza (Nemeth i in., 2004). Regulując żelazo w osoczu i ogólnoustrojową homeostazę żelaza, oś hepcydyna/ferroportyna silnie wpływa na erytropoezę, stąd możliwy rozwój niedokrwistości.
The Iron-Erythropoiesis Connection
Proces wytwarzania czerwonych krwinek zużywa około 80% krążącego żelaza na syntezę hemoglobiny dojrzewających erytroblastów. Większość żelaza (20-25 mg/dobę) jest odzyskiwana przez makrofagi, podczas gdy ograniczona ilość (1-2 mg/dobę) pochodzi z wchłaniania jelitowego. Nerkowy hormon erytropoetyna (EPO) kontroluje proliferację progenitorów erytroidalnych, zwłaszcza CFU-e i w mniejszym stopniu BFU-e, oraz wczesną fazę erytropoezy terminalnej, natomiast zapotrzebowanie na żelazo wzrasta w późnych fazach różnicowania z proerytroblastów do retikulocytów, w celu syntezy hemu włączonego do hemoglobiny (Muckenthaler i wsp., 2017).
Regulacja hepcydyny wymaga crosstalk pomiędzy komórkami zatokowymi śródbłonka wątroby (LSEC), które produkują białka morfogenetyczne kości (BMP) w celu aktywacji szlaku BMP-SMAD, a hepatocytami, które produkują i uwalniają hepcydynę (Babitt i in., 2006; Rausa i in., 2015). BMP6 i BMP2 są najważniejszymi BMP, które podnoszą poziom hepcydyny, podczas gdy ekspresja BMP6 jest zależna od żelaza (Andriopoulos i wsp., 2009; Meynard i wsp., 2009) BMP2 wydaje się mniej reagować na żelazo (Canali i wsp., 2017; Koch i wsp., 2017).
Stopień hepcydyny jest niski w bezwzględnym niedoborze żelaza i niedokrwistości z niedoboru żelaza. W tych warunkach magazyny żelaza są wyczerpane, a sygnalizacja BMP-SMAD jest wyłączona na wielu poziomach. Po pierwsze, ekspresja BMP6 jest tłumiona; następnie aktywność TMPRSS6, proteazy, która rozszczepia hemojuvelinę – ko-receptor BMP (Silvestri i in., 2008), jest silnie zwiększona (Lakhal i in., 2011); i po trzecie, deacetylaza histonowa3 (HDAC3) tłumi locus hepcydyny (Pasricha i in., 2017). W warunkach niedoboru żelaza zmniejszenie produkcji hepcydyny jest mechanizmem adaptacyjnym, który ułatwia dietetyczne i farmakologiczne wchłanianie żelaza (Camaschella i Pagani, 2018).
Gdy niedokrwistość jest ciężka, współistniejąca hipoksja stymuluje erytropoezę poprzez zwiększoną nerkową syntezę i uwalnianie EPO. Prowadzi to do supresji transkrypcji hepcydyny przez erytroferron (ERFE), gen docelowy EPO produkowany przez erytroblasty (Kautz i wsp., 2014), przez cząsteczki (np. PDGF-BB) uwalniane przez inne tkanki (Sonnweber i wsp., 2014) oraz prawdopodobnie przez rozpuszczalne składniki receptorów transferyny (TFR), sTFR1 (Beguin, 2003) i sTFR2 (Pagani i wsp., 2015). Ostatecznym celem jest dostarczenie wystarczającej ilości żelaza na potrzeby rozszerzonej erytropoezy.
Anemias With Abnormal Hepcidin Levels
Anemie mogą być klasyfikowane na podstawie poziomu hepcydyny jako anemie z wysokim i niskim poziomem hepcydyny. Intuicyjne jest, że utrzymujący się wysoki poziom hepcydyny, poprzez blokowanie wchłaniania żelaza, powoduje niedokrwistość z niedoboru żelaza z powodu zmniejszonego dostarczania żelaza do erytropoezy. I odwrotnie, nieefektywna erytropoeza charakteryzuje tzw. niedokrwistości z niedoboru żelaza, w których występuje niski poziom hepcydyny i przeładowanie żelazem. Te dwie grupy niedokrwistości są wynikiem przeciwstawnych mechanizmów patofizjologicznych (ryc. 1). W pierwszej grupie niedokrwistość wynika z hamującego wpływu wywieranego przez hepcydynę na wchłanianie i recykling żelaza, co prowadzi do ogólnoustrojowego niedoboru żelaza; w drugiej grupie niedokrwistość wynika z supresji hepcydyny przez rozszerzoną nieprawidłową erytropoezę (Camaschella i Nai, 2016).
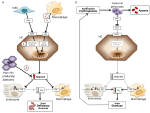
Rysunek 1. Schematyczne przedstawienie mechanizmów niedokrwistości z wysoką (lewy panel) i niską hepcydyną (prawy panel). Panel (A). Molekularna patogeneza niedokrwistości związanej z wysokim poziomem hepcydyny. LSEC, komórki śródbłonka zatok wątrobowych produkujące białka morfogenetyczne kości (BMPs); BMPRs, receptory BMP; IL6, interleukina 6; HC, hepatocyty; HAMP, gen hepcydyny. Fe, żelazo; FPN, ferroportyna; 1, IRIDA; 2, niedokrwistość zapalna; 3, gruczolak produkujący hepcidin. Panel (B). Molekularna patogeneza zmienności hepcydyny w niedokrwistościach spowodowanych nieefektywną erytropoezą. ERFE, erythroferrone sequestering BMPs. Inne mechanizmy hamujące działanie hepcydyny w tym typie niedokrwistości, takie jak zmniejszenie wysycenia transferyny i hipoksja, nie zostały przedstawione. Patrz tekst dla szczegółów.
Anemia Associated With High Hepcidin Levels
Ta grupa obejmuje dwa dziedziczne rzadkie zaburzenia (niedokrwistość z opornego na leczenie niedoboru żelaza i gruczolaki produkujące hepcidynę w wrodzonym błędzie metabolizmu glukozy) oraz nabyte częste schorzenie: niedokrwistość zapalną (Tabela 1).
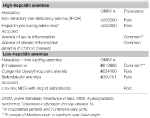
Tabela 1. Niedokrwistości klasyfikowane na podstawie poziomu hepcydyny.
Niedokrwistość z niedoboru żelaza oporna na leczenie
Niedokrwistość z niedoboru żelaza oporna na leczenie (IRIDA) jest rzadkim recesywnym zaburzeniem charakteryzującym się hipochromiczną niedokrwistością mikrocytarną, niskim wysyceniem transferyny i nieprawidłowym prawidłowym/wysokim poziomem hepcydyny. Spowodowane jest mutacjami TMPRSS6 (Finberg i in., 2008), genu kodującego proteazę serynową typu II, matryptazę-2 (Du i in., 2008). Mutacje TMPRSS6 są rozproszone wzdłuż genu i mogą dotyczyć różnych domen, zwłaszcza domeny katalitycznej (De Falco i wsp., 2014). Ta transmembranowa proteaza, ulegająca wysokiej ekspresji w wątrobie, hamuje transkrypcję hepcydyny poprzez rozszczepianie hemojuveliny, współreceptora BMP na powierzchni komórki, hamując w ten sposób sygnalizację BMP i syntezę hepcydyny (Silvestri i in., 2008). Funkcja TMPRSS6 jest niezbędna w niedoborze żelaza, aby umożliwić mechanizm kompensacyjny zwiększonego wchłaniania żelaza.
IRIDA jest obecna od urodzenia i zwykle rozpoznawana w dzieciństwie. W porównaniu z klasycznym niedoborem żelaza, parametry żelaza są nietypowe i nasuwają podejrzenie choroby. Procentowe wysycenie transferyny jest silnie obniżone (poniżej 10%), podobnie jak w innych postaciach niedoboru żelaza; jednakże, w przeciwieństwie do niedoboru żelaza, poziom ferrytyny w surowicy jest prawidłowy/wzrośnięty (Camaschella, 2013; De Falco et al., 2013). Odzwierciedla to zwiększoną akumulację ferrytyny w makrofagach, ze względu na wysoki poziom hepcydyny, która indukuje sklepową sekwestrację żelaza.
Żaden z testów proponowanych do diagnostyki IRIDA nie obejmuje 100% przypadków. Genetyczny test identyfikuje , że TMPRSS6 mutacje, które w niektórych przypadkach (non-sense, mutacje przesunięcia ramki i splicing), są wyraźnie przyczynowe. W innych przypadkach, jak w przypadku wcześniej niezgłaszanych mutacji typu missense, do wykazania przyczynowości potrzebne są badania funkcjonalne (Silvestri i in., 2013). Badania te są jednak rzadko dostępne. Poziom hepcydyny w surowicy jest zwykle podwyższony/normalny, niezależnie od niedoboru żelaza, i zgodny z wysokim/normalnym poziomem ferrytyny. Ważne jest wykluczenie zapalenia przez równoczesne dawkowanie białka C-reaktywnego.
U niektórych pacjentów z fenotypem opornego na leczenie niedoboru żelaza odnotowano obecność pojedynczego zmutowanego allelu TMPRSS6; w tym przypadku debata dotyczy tego, czy należy ich uznać za IRIDA, czy nie. Można przewidzieć spektrum stanów chorobowych, od klasycznego ciężkiego IRIDA spowodowanego homozygotycznymi lub złożonymi heterozygotycznymi mutacjami TMPRSS6 do zwiększonej podatności na niedobór żelaza przyznanej przez pojedyncze mutacje/zmiany polimorficzne. Jednym z podejść proponowanych do przewidywania klasycznego IRIDA jest normalizacja hepcydyny na inne parametry żelaza, jako stosunek saturacji transferyny (Tsat)/log hepcydyny lub Tsat/log ferrytyny (Donker i wsp., 2016). Według innych autorów, większość pacjentów z ciężkim fenotypem IRIDA ma bialleliczne mutacje TMPRSS6, a w przypadku niezidentyfikowania drugi allel może być genetycznie okultystyczny (Heeney i wsp., 2018). Ogólnie rzecz biorąc, osoby z pojedynczym allelem mają łagodniejszy fenotyp niż osoby z dwiema mutacjami i lepiej odpowiadają na leczenie żelazem (Donker i in., 2016). Co ciekawe, wykazano, że kilka SNP TMPRSS6 zapewnia podatność na niedobór żelaza w niektórych populacjach (An i in., 2012) oraz u dawców krwi (Sorensen i in., 2019).
Odnotowano dziedziczenie digeniczne u 5-letniej kobiety, u której pierwotnie stwierdzono nietypowy genotyp IRIDA z jedną mutacją TMPRSS6 (I212T) przyczynową i jedną (R271Q) cichą (De Falco i in., 2014). Później rozpoznano u niej Fibrodysplasia ossificans progressiva (FOP), rzadkie dominujące zaburzenie z ektopowym tworzeniem kości w tkankach miękkich, spowodowane zmutowanym genem receptora BMP typu I ACVR1, kodującym ALK2 (Shore i wsp., 2006). Patologiczny allel ALK2R258S jest konstytutywnie aktywny, ponieważ mutacja dotyczy bogatej w glicynę-serynę domeny genu i sprawia, że szlak BMP/SMAD jest nadaktywny, będąc niezdolnym do wiązania swojego specyficznego inhibitora FKBP12 (Pagani i in., 2017).
Ten rzadki przypadek jest szczególnie ilustracyjny. Po pierwsze, ponieważ domena bogata w glicynę-serynę ALK2 oddziałuje z FKBP12, a mutacja destabilizuje to wiązanie, ujawniła wcześniej niepodejrzewaną rolę FKBP12 jako modulatora wątrobowej ALK2 i hepcydyny (Colucci i in., 2017). Po drugie, doprowadziło do zidentyfikowania związku pomiędzy aktywacją kostnych i wątrobowych receptorów BMP typu I. Po trzecie, przypadek ten wzmacnia znaczenie nienaruszonego TMPRSS6 w kontrolowaniu wątrobowej sygnalizacji BMP/SMAD, ponieważ nie zidentyfikowano IRIDA wśród innych pacjentów FOP z tą samą mutacją ACVR1 i przypuszczalnie normalnym TMPRSS6 (Pagani i in., 2017). Wreszcie, ten przypadek jest zgodny z koncepcją, że haploinsufficiency TMPRSS6 nie może powodować klasycznego IRIDA.
Optymalne leczenie IRIDA jest nieokreślone. Doustne podawanie żelaza jest nieskuteczne, ponieważ nie jest ono wchłaniane. Dodatek witaminy C pozwala na sporadyczną odpowiedź. Żelazo podawane dożylnie wywołuje częściową odpowiedź, zwykle w wolniejszym tempie w porównaniu z pacjentami z nabytym niedoborem żelaza. EPO jest nieskuteczna w klasycznych przypadkach (De Falco i wsp., 2013; Heeney i Finberg, 2014).
Anemia of Hepcidin-Producing Adenomas
Jest to niezwykle rzadkie schorzenie występujące u dorosłych pacjentów dotkniętych chorobą spichrzeniową glikogenu 1a, recesywnym zaburzeniem spowodowanym niedoborem fosfatazy glukozo-6, która katalizuje reakcję biorącą udział zarówno w glikogenolizie, jak i glukoneogenezie. Częstym, niebezpiecznym objawem choroby jest hipoglikemia. Obecne leczenie prowadzi do wydłużenia czasu przeżycia chorych dzieci do wieku dorosłego, przy jednoczesnym występowaniu szeregu powikłań, takich jak niedokrwistość i gruczolaki wątroby. Niedokrwistość jest mikrocytarna i hipochromiczna, z niedoborem żelaza, oporna na doustne leczenie żelazem. Niedokrwistość powróciła po chirurgicznym wycięciu gruczolaka. W tkance gruczolaka stwierdzono pozytywny wynik mRNA dla hepcydyny, podczas gdy prawidłowa tkanka otaczająca wykazywała supresję hepcydyny, co było oczekiwane z powodu ektopowej niekontrolowanej produkcji hepcydyny (Weinstein et al., 2002). Cechy hematologiczne pacjentów przypominają te z IRIDA, ponieważ dzielą oni wysokie poziomy hepcydyny jako wspólny mechanizm niedokrwistości.
Anemia of Inflammation
Anemia of inflammation (AI), wcześniej znana jako niedokrwistość chorób przewlekłych, jest umiarkowaną niedokrwistością normochromowo-normocytową, która rozwija się w warunkach ogólnoustrojowego zapalenia i aktywacji immunologicznej. Występuje w kilku powszechnych schorzeniach, w tym w przewlekłych infekcjach, chorobach autoimmunologicznych, zaawansowanej chorobie nowotworowej, przewlekłej chorobie nerek, zastoinowej niewydolności serca, przewlekłej obturacyjnej chorobie płuc, niedokrwistości osób starszych (przynajmniej częściowo) oraz chorobie przeszczep przeciwko gospodarzowi. AI jest jedną z najczęstszych niedokrwistości na świecie i najczęstszą niedokrwistością u pacjentów hospitalizowanych. Ostry stan zapalny przyczynia się do nasilenia niedokrwistości na oddziałach intensywnej terapii. Mechanizmy molekularne leżące u podłoża AI są wielorakie i złożone. Nadprodukcja cytokin, takich jak IL1-β, TNF-α i IL-6 przez makrofagi oraz INF-γ przez limfocyty, hamuje produkcję EPO, upośledza odpowiedź erytropoezy, zwiększa stężenie hepcydyny i może aktywować erytrofagocytozę, zwłaszcza w ostrych postaciach (Weiss i Goodnough, 2005; Ganz, 2019).
Hepcydyna jest aktywowana przez IL-6 poprzez receptor IL-6 (IL-6R) i sygnalizację JAK2-STAT3. Pełna aktywacja hepcydyny wymaga aktywnego szlaku BMP-SMAD, ponieważ inaktywacja sygnalizacji BMP zmniejsza hepcydynę w zwierzęcych modelach zapalenia (Theurl et al., 2011). Rozregulowanie ogólnoustrojowej homeostazy żelaza powoduje sekwestrację żelaza przez makrofagi oraz zmniejszone wchłanianie i recykling, co prowadzi do niskiego nasycenia transferyny i ograniczenia żelaza w erytropoezie i innych tkankach.
Tradycyjne leczenie AI opiera się na odwracalności/kontroli choroby podstawowej, gdy tylko jest to możliwe. Jeśli choroba nie jest uleczalna, a niedokrwistość jest łagodna, konieczna jest staranna ocena stosunku korzyści do ryzyka, aby uniknąć działań niepożądanych jakiegokolwiek leczenia. Leczenie oparte na patofizjologii jest ograniczone do stosowania związków podobnych do erytropoetyny i żelaza. Stosowanie czynników stymulujących erytropoezę (ESA) powoduje supresję hepcydyny poprzez indukcję ekspansji erytropoezy. Takie postępowanie jest szeroko stosowane u pacjentów z przewlekłą chorobą nerek, zespołami mielodysplastycznymi niskiego ryzyka oraz u chorych na nowotwory poddawanych chemioterapii. Konieczna jest jednak staranna kontrola kliniczna, ponieważ duże dawki mają działania niepożądane ze strony układu sercowo-naczyniowego. Podawanie żelaza dożylnie może złagodzić niedobór żelaza spowodowany zależną od ESA ekspansją erytropoezy. Podawanie żelaza doustnie jest zwykle nieskuteczne, ponieważ wysoki poziom hepcydyny przeciwdziała jego wchłanianiu jelitowemu. Inhibitory hydroksylazy prolylowej (hypoxia inducible factor, HIF stabilizatory) są eksperymentalnie stosowane w przewlekłej chorobie nerek w celu zwiększenia endogennej EPO. Przewlekłe leczenie transfuzjami krwinek czerwonych nie jest zalecane ze względu na przemijający efekt i działania niepożądane; ogranicza się je do ciężkiej opornej niedokrwistości (Camaschella, 2019; Weiss et al., 2019).
Anemias Associated With Low Hepcidin Levels
Nieefektywna erytropoeza i niskie lub nieprawidłowo prawidłowe stężenie hepcydyny, z następczym przeładowaniem żelazem, są typowymi cechami niedokrwistości „obciążających żelazem”.” Prototypem jest β-talasemia, genetyczna choroba recesywna spowodowana mutacjami genu β-globiny, które powodują niedokrwistość i nadmierną produkcję łańcucha α-globiny. Ten ostatni wytrąca się w szpiku kostnym w postaci hemichromów, uszkadzając dojrzewające prekursory erytroidów i prowadząc do nieefektywnej erytropoezy. Dzieje się tak w talasemii nie zależnej od transfuzji lub talasemii pośredniej, w której erytropoeza charakteryzuje się przewagą niedojrzałych komórek, które uwalniając erytroferron hamują ekspresję wątrobowej hepcydyny. Poziom hepcydyny jest zwykle większy w talasemii zależnej od transfuzji, w której endogenna nieefektywna erytropoeza jest przynajmniej częściowo tłumiona przez transfuzje (Camaschella i Nai, 2016).
W supresji hepcydyny pośredniczy zwiększona ilość cytokiny erytroferronu (ERFE), członka rodziny TNF-α kodowanego przez gen ERFE, syntetyzowanego przez erytroblasty po stymulacji EPO (Kautz i wsp., 2014). ERFE jest uwalniany do krążenia i sekwestruje BMP, zwłaszcza BMP6 (Arezes i wsp., 2018), osłabiając sygnalizację hepcydyny w odpowiedzi na żelazo. Dodatkowo w locus hepcydyny dochodzi do epigenetycznej supresji przez deacetylazę histonową HDAC3 (Pasricha i wsp., 2017). Gdy niedokrwistość powoduje niedotlenienie, dochodzi do wydzielania innych mediatorów, takich jak PDGF-BB (Sonnweber i wsp., 2014), który jest uwalniany przez różne typy komórek, tłumią hepcidin.
Stopień hepcydyny jest obniżony przez specjalny mechanizm w mielodysplazji niskiego ryzyka z obrączkowanymi sideroblastami, zaburzeniu klonalnym spowodowanym mutacjami genu spliceosomu SF3B1. Żelazo gromadzi się w mitochondriach, prowadząc do nieefektywnej erytropoezy i ogólnoustrojowego przeciążenia żelazem. Nieprawidłowo splicowane, wydłużone białko ERFE jest silniejsze niż ERFE typu dzikiego w supresji hepcydyny (Bondu i wsp., 2019) i powodowaniu niezależnego od transfuzji obciążenia żelazem.
Targeted Therapies for Hepcidin-Related Anemias
Rozpoznanie mechanizmów molekularnych odpowiedzialnych za omawiane wcześniej niedokrwistości pobudziło badania nad opracowaniem terapii celowanych, które miałyby zastąpić obecne leczenie objawowe (Sebastiani et al., 2016; Crielaard et al., 2017). Podejścia różnią się w zależności od rodzaju niedokrwistości i celu, jakim jest obniżenie lub podwyższenie poziomu hepcydyny lub ich działania (Tabela 2).
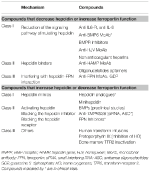
Tabela 2. Experimental therapies targeting the hepcidin-ferroportin axis.
Experimental Therapies to Decrease Hepcidin Levels/Increase Ferroportin Function
Z wyjątkiem guzów produkujących hepcidinę, które muszą być usuwane chirurgicznie, związki antagonizujące hepcidinę lub jej działanie mogą być użyteczne we wszystkich niedokrwistościach charakteryzujących się wysokim poziomem hepcidiny. Ich główne zastosowanie znalazłoby się w przewlekłych chorobach zapalnych w celu odwrócenia hipoferremii i niedokrwistości. Kilka eksperymentalnych terapii mających na celu manipulowanie szlakiem hepcydyny i jej funkcją zostało przebadanych w badaniach przedklinicznych. Antagoniści hepcydyny to inhibitory syntezy/regulatorów hepcydyny (Ganz, 2019), związki wiążące hepcydynę, które blokują jej funkcję, oraz związki zaburzające interakcję hepcydyna-ferroportyna (tab. 2). Niektóre związki są w fazie badań klinicznych szczególnie w przewlekłej chorobie nerek (Sheetz et al., 2019). W IRIDA manipulacja szlakiem hepcydyny została zaproponowana w badaniach przedklinicznych z zastosowaniem anty-HJV MoAb (Kovac et al., 2016).
Experimental Therapies to Increase Hepcidin Levels/Decrease Ferroportin Function
Zwiększenie poziomu hepcydyny może nie tylko zmniejszyć przeciążenie żelazem, ale również częściowo kontrolować nieefektywną erytropoezę w niedokrwistościach obciążonych żelazem. Wśród tych schorzeń najlepiej przebadana jest β-talasemia (Casu et al., 2018; Gupta et al., 2018). Proponowane leki to analogi hepcydyny (niektóre w badaniach klinicznych), modulatory hepcydyny, zwłaszcza inhibitory TMPRSS6, lub związki zaburzające interakcję hepcydyna-ferroportyna zmniejszające eksport żelaza (tab. 2).
Podczas gdy związki zwiększające stężenie hepcydyny zmniejszają nieefektywną erytropoezę ze względu na błędne koło pomiędzy nieefektywną erytropoezą a obciążeniem żelazem (Camaschella i Nai, 2016), leki sprzyjające dojrzewaniu prekursorów erytroidów, jak pułapka ligandowa receptora aktywiny IIB, luspatercept, nie tylko poprawiają niedokrwistość, ale również polepszają homeostazę żelaza poprzez zmniejszenie hamowania hepcydyny (Piga i in., 2019).
Kilka ukierunkowanych podejść znajdujących się obecnie w badaniach klinicznych, miejmy nadzieję, zaowocuje nowatorskimi metodami leczenia różnych niedokrwistości.
Podsumowanie
Spektakularne postępy w zrozumieniu regulacji metabolizmu żelaza i hepcydyny pozwoliły na lepsze zrozumienie kontroli erytropoezy, ponieważ wraz z erytropoetyną żelazo jest podstawowym czynnikiem dojrzewania komórek erytroidalnych. Stany prowadzące do anemii mogą być związane z wysokim lub niskim poziomem hepcydyny. W obu przypadkach, kontrastowa deregulacja hepcydyny może poprawić/usunąć niedokrwistość w modelach przedklinicznych, oferując nowe narzędzia, które już są lub wkrótce będą badane klinicznie w leczeniu konkretnych niedokrwistości.
Author Contributions
AP przygotował projekt pracy. CC opracowała ostateczną wersję. AN i LS przyczynili się do napisania i krytycznej recenzji manuskryptu. Wszyscy autorzy zatwierdzili ostateczną wersję.
Funding
Ta praca była wspierana częściowo przez EHA Advanced Research Grant w 2018 roku do AP. Cariplo Foundation Young Investigator Grant n° 2017-0916 do AN.
Konflikt interesów
CC jest konsultantem Vifor Pharma, Celgene i Novartis.
Pozostali autorzy deklarują, że badania były prowadzone przy braku jakichkolwiek komercyjnych lub finansowych relacji, które mogłyby być interpretowane jako potencjalny konflikt interesów.
An, P., Wu, Q., Wang, H., Guan, Y., Mu, M., Liao, Y., et al. (2012). TMPRSS6, ale nie TF, TFR2 lub BMP2 variants are associated with increased risk of iron-deficiency anemia. Hum. Mol. Genet. 21, 2124-2131. doi: 10.1093/hmg/dds028
PubMed Abstract | CrossRef Full Text | Google Scholar
Andriopoulos, B. Jr., Corradini, E., Xia, Y., Faasse, S. A., Chen, S., Grgurevic, L., et al. (2009). BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat. Genet. 41, 482-487. doi: 10.1038/ng.335
PubMed Abstract | CrossRef Full Text | Google Scholar
Arezes, J., Foy, N., McHugh, K., Sawant, A., Quinkert, D., Terraube, V., et al. (2018). Erythroferrone hamuje indukcję hepcydyny przez BMP6. Blood 132, 1473-1477. doi: 10.1182/blood-2018-06-857995
PubMed Abstract | CrossRef Full Text | Google Scholar
Aschemeyer, S., Qiao, B., Stefanova, D., Valore, E. V., Sek, A. C., Ruwe, T. A., et al. (2018). Structure-function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin. Blood. 131, 899-910.doi: 10.1182/blood-2017-05-786590
CrossRef Full Text | Google Scholar
Babitt, J. L., Huang, F. W., Wrighting, D. M., Xia, Y., Sidis, Y., Samad, T. A., et al. (2006). Bone morphogenetic protein signaling by hemojuvelin reguluje ekspresję hepcidin. Nat. Genet. 38, 531-539. doi: 10.1038/ng1777
PubMed Abstract | CrossRef Full Text | Google Scholar
Beguin, Y. (2003). Soluble transferrin receptor for the evaluation of erythropoiesis and iron status. Clin. Chim. Acta 329, 9-22. doi: 10.1016/S0009-8981(03)00005-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Bondu, S., Alary, A. S., Lefevre, C., Houy, A., Jung, G., Lefebvre, T., et al. (2019). Wariant erytroferronu zaburza homeostazę żelaza w zmutowanym SF3B1 zespole mielodysplastycznym. Sci. Transl. Med. 11:pii:eaav5467. doi: 10.1126/scitranslmed.aav5467
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C. (2013). Jak zarządzam pacjentami z atypową niedokrwistością mikrocytarną. Br. J. Haematol. 160, 12-24. doi: 10.1111/bjh.12081
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C. (2019). Niedobór żelaza. Blood 133, 30-39. doi: 10.1182/blood-2018-05-815944
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C., and Nai, A. (2016). Ineffective erythropoiesis and regulation of iron status in iron loading anaemias. Br. J. Haematol. 172, 512-523. doi: 10.1111/bjh.13820
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C., and Pagani, A. (2018). Advances in understanding iron metabolism and its crosstalk with erythropoiesis. Br. J. Haematol. 182, 481-494. doi: 10.1111/bjh.15403
PubMed Abstract | CrossRef Full Text | Google Scholar
Canali, S., Wang, C. Y., Zumbrennen-Bullough, K. B., Bayer, A., and Babitt, J. L. (2017). Bone morphogenetic protein 2 controls iron homeostasis in mice independent of Bmp6. Am. J. Hematol. 92, 1204-1213. doi: 10.1002/ajh.24888
PubMed Abstract | CrossRef Full Text | Google Scholar
Casu, C., Nemeth, E., and Rivella, S. (2018). Agoniści hepcydyny jako narzędzia terapeutyczne. Blood 131, 1790-1794. doi: 10.1182/blood-2017-11-737411
PubMed Abstract | CrossRef Full Text | Google Scholar
Colucci, S., Pagani, A., Pettinato, M., Artuso, I., Nai, A., Camaschella, C., et al. (2017). Immunofilina FKBP12 hamuje ekspresję hepcydyny poprzez wiązanie receptora BMP typu I ALK2 w hepatocytach. Blood 130, 2111-2120. doi: 10.1182/blood-2017-04-780692
PubMed Abstract | CrossRef Full Text | Google Scholar
Crielaard, B. J., Lammers, T., and Rivella, S. (2017). Targeting iron metabolism in drug discovery and delivery. Nat. Rev. Drug Discov. 16, 400-423. doi: 10.1038/nrd.2016.248
PubMed Abstract | CrossRef Full Text | Google Scholar
De Falco, L., Sanchez, M., Silvestri, L., Kannengiesser, C., Muckenthaler, M. U., Iolascon, A., et al. (2013). Iron refractory iron deficiency anemia. Haematologica 98, 845-853. doi: 10.3324/haematol.2012.075515
PubMed Abstract | CrossRef Full Text | Google Scholar
De Falco, L., Silvestri, L., Kannengiesser, C., Moran, E., Oudin, C., Rausa, M., et al. (2014). Functional and clinical impact of novel TMPRSS6 variants in iron-refractory iron-deficiency anemia patients and genotype-phenotype studies. Hum. Mutat. 35, 1321-1329. doi: 10.1002/humu.22632
PubMed Abstract | CrossRef Full Text | Google Scholar
Donker, A. E., Schaap, C. C., Novotny, V. M., Smeets, R., Peters, T. M., van den Heuvel, B. L., et al. (2016). Iron refractory iron deficiency anemia: a heterogeneous disease that is not always iron refractory. Am. J. Hematol. 91, E482-E490. doi: 10.1002/ajh.24561
PubMed Abstract | CrossRef Full Text | Google Scholar
Du, X., She, E., Gelbart, T., Truksa, J., Lee, P., Xia, Y., et al. (2008). The serine protease TMPRSS6 is required to sense iron deficiency. Science 320, 1088-1092. doi: 10.1126/science.1157121
PubMed Abstract | CrossRef Full Text | Google Scholar
Finberg, K. E., Heeney, M. M., Campagna, D. R., Aydinok, Y., Pearson, H. A., Hartman, K. R., et al. (2008). Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA). Nat. Genet. 40, 569-571. doi: 10.1038/ng.130
PubMed Abstract | CrossRef Full Text | Google Scholar
Ganz, T. (2019). Niedokrwistość z zapalenia. N. Engl. J. Med. 381, 1148-1157. doi: 10.1056/NEJMra1804281
PubMed Abstract | CrossRef Full Text | Google Scholar
Gupta, R., Musallam, K. M., Taher, A. T., and Rivella, S. (2018). Nieefektywna erytropoeza: niedokrwistość i przeciążenie żelazem. Hematol. Oncol. Clin. North Am. 32, 213-221. doi: 10.1016/j.hoc.2017.11.009
PubMed Abstract | CrossRef Full Text | Google Scholar
Heeney, M. M., and Finberg, K. E. (2014). Niedokrwistość z niedoboru żelaza oporna na leczenie (IRIDA). Hematol. Oncol. Clin. North Am. 28, 637-652. doi: 10.1016/j.hoc.2014.04.009
PubMed Abstract | CrossRef Full Text | Google Scholar
Heeney, M. M., Guo, D., De Falco, L., Campagna, D. R., Olbina, G., Kao, P. P., et al. (2018). Normalizacja hepcydyny przewiduje status mutacji TMPRSS6 u pacjentów z przewlekłym niedoborem żelaza. Blood 132, 448-452. doi: 10.1182/blood-2017-03-773028
PubMed Abstract | CrossRef Full Text | Google Scholar
Kautz, L., Jung, G., Valore, E. V., Rivella, S., Nemeth, E., and Ganz, T. (2014). Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 46, 678-684. doi: 10.1038/ng.2996
PubMed Abstract | CrossRef Full Text | Google Scholar
Koch, P. S., Olsavszky, V., Ulbrich, F., Sticht, C., Demory, A., Leibing, T., et al. (2017). Angiocrine Bmp2 signaling in murine liver controls normal iron homeostasis. Blood 129, 415-419. doi: 10.1182/blood-2016-07-729822
PubMed Abstract | CrossRef Full Text | Google Scholar
Kovac, S., Boser, P., Cui, Y., Ferring-Appel, D., Casarrubea, D., Huang, L., et al. (2016). Anti-hemojuvelin antibody corrects anemia caused by inappropriately high hepcidin levels. Haematologica 101, e173-e176. doi: 10.3324/haematol.2015.140772
PubMed Abstract | CrossRef Full Text | Google Scholar
Lakhal, S., Schodel, J., Townsend, A. R., Pugh, C. W., Ratcliffe, P. J., and Mole, D. R. (2011). Regulation of type II transmembrane serine proteinase TMPRSS6 by hypoxia-inducible factors: new link between hypoxia signaling and iron homeostasis. J. Biol. Chem. 286, 4090-4097. doi: 10.1074/jbc.M110.173096
PubMed Abstract | CrossRef Full Text | Google Scholar
Meynard, D., Kautz, L., Darnaud, V., Canonne-Hergaux, F., Coppin, H., and Roth, M. P. (2009). Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat. Genet. 41, 478-481. doi: 10.1038/ng.320
PubMed Abstract | CrossRef Full Text | Google Scholar
Muckenthaler, M. U., Rivella, S., Hentze, M. W., and Galy, B. (2017). Czerwony dywan dla metabolizmu żelaza. Cell 168, 344-361. doi: 10.1016/j.cell.2016.12.034
PubMed Abstract | CrossRef Full Text | Google Scholar
Nemeth, E., Tuttle, M. S., Powelson, J., Vaughn, M. B., Donovan, A., Ward, D. M., et al. (2004). Hepcidin reguluje komórkowy odpływ żelaza poprzez wiązanie się z ferroportyną i indukowanie jej internalizacji. Science 306, 2090-2093. doi: 10.1126/science.1104742
PubMed Abstract | CrossRef Full Text | Google Scholar
Pagani, A., Colucci, S., Bocciardi, R., Bertamino, M., Dufour, C., Ravazzolo, R., et al. (2017). A new form of IRIDA due to combined heterozygous mutations of TMPRSS6 and ACVR1A encoding the BMP receptor ALK2. Blood 129, 3392-3395. doi: 10.1182/blood-2017-03-773481
PubMed Abstract | CrossRef Full Text | Google Scholar
Pagani, A., Vieillevoye, M., Nai, A., Rausa, M., Ladli, M., Lacombe, C., et al. (2015). Regulation of cell surface transferrin receptor-2 by iron-dependent cleavage and release of a soluble form. Haematologica 100, 458-465. doi: 10.3324/haematol.2014.118521
PubMed Abstract | CrossRef Full Text | Google Scholar
Pasricha, S. R., Lim, P. J., Duarte, T. L., Casu, C., Oosterhuis, D., Mleczko-Sanecka, K., et al. (2017). Hepcidin is regulated by promoter-associated histone acetylation and HDAC3. Nat. Commun. 8:403. doi: 10.1038/s41467-017-00500-z
PubMed Abstract | CrossRef Full Text | Google Scholar
Piga, A., Perrotta, S., Gamberini, M. R., Voskaridou, E., Melpignano, A., Filosa, A., et al. (2019). Luspatercept poprawia poziom hemoglobiny i wymagania dotyczące transfuzji krwi w badaniu pacjentów z beta-talasemią. Blood 133, 1279-1289. doi: 10.1182/blood-2018-10-879247
PubMed Abstract | CrossRef Full Text | Google Scholar
Rausa, M., Pagani, A., Nai, A., Campanella, A., Gilberti, M. E., Apostoli, P., et al. (2015). Bmp6 expression in murine liver non parenchymal cells: a mechanism to control their high iron exporter activity and protect hepatocytes from iron overload? PLoS One 10:e0122696. doi: 10.1371/journal.pone.0122696
PubMed Abstract | CrossRef Full Text | Google Scholar
Sebastiani, G., Wilkinson, N., and Pantopoulos, K. (2016). Pharmacological targeting of the hepcidin/ferroportin axis. Front. Pharmacol. 7:160. doi: 10.3389/fphar.2016.00160
PubMed Abstract | CrossRef Full Text | Google Scholar
Sheetz, M., Barrington, P., Callies, S., Berg, P. H., McColm, J., Marbury, T., et al. (2019). Targeting the hepcidin-ferroportin pathway in anaemia of chronic kidney disease. Br. J. Clin. Pharmacol. 85, 935-948. doi: 10.1111/bcp.13877
PubMed Abstract | CrossRef Full Text | Google Scholar
Shore, E. M., Xu, M., Feldman, G. J., Fenstermacher, D. A., Cho, T. J., Choi, I. H., et al. (2006). A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 38, 525-527. doi: 10.1038/ng1783
PubMed Abstract | CrossRef Full Text | Google Scholar
Silvestri, L., Pagani, A., Nai, A., De Domenico, I., Kaplan, J., and Camaschella, C. (2008). The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. 8, 502-511. doi: 10.1016/j.cmet.2008.09.012
PubMed Abstract | CrossRef Full Text | Google Scholar
Silvestri, L., Rausa, M., Pagani, A., Nai, A., and Camaschella, C. (2013). How to assess causality of TMPRSS6 mutations? Hum. Mutat. 34, 1043-1045. doi: 10.1002/humu.22321
PubMed Abstract | CrossRef Full Text | Google Scholar
Sonnweber, T., Nachbaur, D., Schroll, A., Nairz, M., Seifert, M., Demetz, E., et al. (2014). Hypoxia induced downregulation of hepcidin is mediated by platelet derived growth factor BB. Gut 63, 1951-1959. doi: 10.1136/gutjnl-2013-305317
PubMed Abstract | CrossRef Full Text | Google Scholar
Sorensen, E., Rigas, A. S., Didriksen, M., Burgdorf, K. S., Thorner, L. W., Pedersen, O. B., et al. (2019). Czynniki genetyczne wpływające na poziom hemoglobiny u 15 567 dawców krwi: wyniki z duńskiego badania dawców krwi. Transfusion 59, 226-231. doi: 10.1111/trf.15075
CrossRef Full Text | Google Scholar
Theurl, I., Schroll, A., Sonnweber, T., Nairz, M., Theurl, M., Willenbacher, W., et al. (2011). Pharmacologic inhibition of hepcidin expression reverses anemia of chronic inflammation in rats. Blood 118, 4977-4984. doi: 10.1182/blood-2011-03-345066
PubMed Abstract | CrossRef Full Text | Google Scholar
Weinstein, D. A., Roy, C. N., Fleming, M. D., Loda, M. F., Wolfsdorf, J. I., and Andrews, N. C. (2002). Inappropriate expression of hepcidin is associated with iron refractory anemia: implications for the anemia of chronic disease. Blood 100, 3776-3781. doi: 10.1182/blood-2002-04-1260
PubMed Abstract | CrossRef Full Text | Google Scholar
Weiss, G., Ganz, T., and Goodnough, L. T. (2019). Anemia of inflammation. Blood 133, 40-50. doi: 10.1182/blood-2018-06-856500
PubMed Abstract | CrossRef Full Text | Google Scholar
Weiss, G., and Goodnough, L. T. (2005). Niedokrwistość w chorobach przewlekłych. N. Engl. J. Med. 352, 1011-1023. doi: 10.1056/NEJMra041809
PubMed Abstract | CrossRef Full Text | Google Scholar
.
Leave a Reply