Frontiers in Physiology
Introduktion
Anemi är en av de vanligaste sjukdomarna i världen och anemi på grund av järnbrist är den vanligaste formen enligt flera analyser (översikt i Camaschella, 2019). Denna typ av anemi beror på järnbrist i hela kroppen och oförmågan att tillföra den stora mängd järn som benmärgen förbrukar för att producera ett tillräckligt antal röda blodkroppar för att upprätthålla syresättningen av vävnaden.
Järntillgängligheten styrs av leverpeptidhormonet hepcidin. Kroppens järnökning orsakar produktion av hepcidin, som frigörs i cirkulationen och verkar på sin receptor ferroportin, ett transmembranärt järnexportörprotein som är starkt uttryckt på enterocyter, makrofager och hepatocyter. Hepcidin minskar järninträdet till plasma från absorberande duodenalceller och järnåtervinande makrofager genom att blockera järnexport (Aschemeyer et al., 2018) och genom att bryta ned järnexportören ferroportin (Nemeth et al., 2004). Genom att reglera plasmajärn och systemisk järnhomeostas påverkar hepcidin/ferroportin-axeln starkt erytropoesin, och därmed den möjliga utvecklingen av anemi.
Järn-Erythropoesis Connection
Processen för produktion av röda blodkroppar förbrukar cirka 80 % av det cirkulerande järnet för hemoglobinsyntesen av mognande erytroblaster. Det mesta järnet (20-25 mg/dag) återvinns av makrofager, medan en begränsad mängd (1-2 mg dagligen) härrör från intestinal absorption. Njurhormonet erytropoietin (EPO) kontrollerar proliferationen av erytroida progenitorer, särskilt CFU-e och i lägre grad BFU-e, och den tidiga fasen av den terminala erytropoesen, medan järnbehovet ökar i de sena differentieringsfaserna från proerytroblaster till retikulocyter, för syntesen av hem som inkorporeras i hemoglobin (Muckenthaler et al, 2017).
Hepcidinreglering kräver en crosstalk mellan leverendoteliala sinusoidala celler (LSEC) som producerar benmorfogenetiska proteiner (BMP) för att aktivera BMP-SMAD-vägen och hepatocyter som producerar och frisätter hepcidin (Babitt et al., 2006; Rausa et al., 2015). BMP6 och BMP2 är de viktigaste BMP:erna som uppreglerar hepcidin, medan BMP6-uttrycket är järnberoende (Andriopoulos et al., 2009; Meynard et al., 2009) BMP2 verkar mindre järnresponsivt (Canali et al., 2017; Koch et al., 2017).
Hepcidinnivåerna är låga vid absolut järnbrist och järnbristanemi. Under dessa förhållanden är järnlagren uttömda och BMP-SMAD-signalering stängs av på flera nivåer. För det första undertrycks BMP6-uttrycket, därefter ökar aktiviteten hos TMPRSS6, ett proteas som klyver BMP-koreceptorn hemojuvelin (Silvestri et al., 2008), kraftigt (Lakhal et al., 2011) och för det tredje undertrycker histondeacetylas3 (HDAC3) hepcidinlocus (Pasricha et al., 2017). Vid förhållanden med järnbrist är minskningen av hepcidinproduktionen en anpassningsmekanism som underlättar absorptionen av järn via kosten och farmakologiskt (Camaschella och Pagani, 2018).
När anemin är allvarlig stimulerar den samtidigt existerande hypoxi erytropoesin genom ökad njursyntes och frisättning av EPO. Detta leder till suppression av hepcidintranskriptionen genom erytroferron (ERFE), en EPO-målgen som produceras av erytroblaster (Kautz et al., 2014), genom molekyler (t.ex. PDGF-BB) som frigörs av andra vävnader (Sonnweber et al., 2014) och troligen av lösliga komponenter av transferrinreceptorer (TFR), sTFR1 (Beguin, 2003) och sTFR2 (Pagani et al., 2015). Det slutliga målet är att tillföra tillräckligt med järn för behoven hos en expanderad erytropoes.
Anemier med onormala hepcidinnivåer
Anemier kan klassificeras på grundval av hepcidinnivåerna som anemier med högt och lågt hepcidin. Det är intuitivt att ihållande höga hepcidinnivåer, genom att blockera järnabsorptionen, orsakar järnbristanemi på grund av minskad järntillförsel till erytropoesin. Omvänt karakteriserar ineffektiv erytropoesi de så kallade järnbelastningsanemier som har låga hepcidinnivåer och järnöverbelastning. Dessa två grupper av anemier är resultatet av motsatta patofysiologiska mekanismer (figur 1). I den första gruppen beror anemin på den hämmande effekt som hepcidin utövar på järnabsorption och järnåtervinning som leder till systemisk järnbrist; i den andra gruppen beror anemin på hepcidinundertryckning genom en expanderad onormal erytropoes (Camaschella och Nai, 2016).
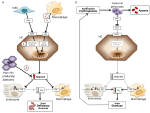
Figur 1. Schematisk representation av mekanismerna för anemier med högt (vänster panel) och lågt hepcidin (höger panel). Panel (A). Molekylär patogenes av anemi associerad med höga hepcidinnivåer. LSEC, leversinusoidala endotelceller som producerar benmorfogenetiska proteiner (BMP); BMPRs, BMP-receptorer; IL6, interleukin 6; HC, hepatocyter; HAMP, hepcidin-gen. Fe, järn; FPN, ferroportin; 1, IRIDA; 2, inflammationsanemi; 3, hepcidinproducerande adenom. Panel (B). Molekylär patogenes av hepcidinvariation vid anemier på grund av ineffektiv erytropoesi. ERFE, erytroferron som binder BMP:er. Andra mekanismer som hämmar hepcidin i denna typ av anemi, som minskad transferrinmättnad och hypoxi, visas inte. Se texten för detaljer.
Ansamling associerad med höga hepcidinnivåer
Denna grupp innefattar två ärftliga sällsynta sjukdomar (järnrefraktär järnbristanemi och hepcidinproducerande adenom i ett medfött fel i glukosmetabolismen) och ett förvärvat vanligt tillstånd: inflammationsanemi (tabell 1).
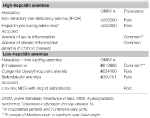
Tabell 1. Anemier klassificerade enligt hepcidinnivåer.
Järnrefraktär järnbristanemi
Järnrefraktär järnbristanemi (IRIDA) är en sällsynt recessiv sjukdom som kännetecknas av hypokrom mikrocytisk anemi, låg transferrinmättnad och olämpligt normala/höga hepcidinnivåer. Den orsakas av mutationer i TMPRSS6 (Finberg et al., 2008), en gen som kodar för typ II-serinproteaset matriptas-2 (Du et al., 2008). Mutationer i TMPRSS6 är spridda längs genen och kan påverka olika domäner, särskilt den katalytiska domänen (De Falco et al., 2014). Detta transmembranproteas, som uttrycks starkt i levern, hämmar hepcidintranskriptionen genom att klyva cellytans BMP-koreceptor hemojuvelin, vilket dämpar BMP-signalering och hepcidinsyntes (Silvestri et al., 2008). TMPRSS6-funktionen är väsentlig vid järnbrist för att möjliggöra den kompensatoriska mekanismen med ökat järnupptag.
IRIDA är närvarande sedan födseln och diagnostiseras vanligen i barndomen. Jämfört med klassisk järnbrist är järnparametrarna atypiska och väcker misstanke om sjukdomen. Den procentuella mättnaden av transferrin är starkt reducerad (mindre än 10 %) som vid andra former av järnbrist; till skillnad från järnbrist är dock nivåerna av serumferritin normala/förhöjda (Camaschella, 2013; De Falco et al., 2013). Detta återspeglar en ökad ferritinackumulering i makrofager, på grund av de höga hepcidinnivåerna som inducerar lagringsjärnsekvestrering.
Ingen av de tester som föreslås för IRIDA-diagnosen täcker 100 % av fallen. Det genetiska testet identifierar att TMPRSS6-mutationer, som i vissa fall (non-sense-, frame-shift- och splicingmutationer), är klart kausala. I andra fall, som i fråga om tidigare oanmälda missense-mutationer, behövs funktionella studier för att påvisa kausalitet (Silvestri et al., 2013). Dessa tester är dock knappt tillgängliga. Serumhepcidinnivåerna är vanligen förhöjda/normala, oberoende av järnbrist, och förenliga med högt/normalt ferritin. Det är viktigt att utesluta inflammation genom att samtidigt dosera C-reaktivt protein.
Vissa patienter med en fenotyp av refraktär järnbrist har rapporterats ha en enda TMPRSS6-muterad allel; här är diskussionen om huruvida de ska betraktas som IRIDA eller inte. Man kan tänka sig ett spektrum av tillstånd som sträcker sig från klassisk svår IRIDA på grund av homozygota eller sammansatta heterozygota TMPRSS6-mutationer till ökad känslighet för järnbrist på grund av enstaka mutationer/polymorfa förändringar. Ett tillvägagångssätt som föreslås för att förutsäga klassisk IRIDA är hepcidin-normalisering på andra järnparametrar, som förhållandet transferrinmättnad (Tsat)/log hepcidin eller Tsat/log ferritin (Donker et al., 2016). Enligt andra författare har de flesta patienter med en allvarlig IRIDA-fenotyp bialleliska TMPRSS6-mutationer och när de inte identifieras kan den andra allelen vara genetiskt ockult (Heeney et al., 2018). Generellt sett har personer med en enda allel en mildare fenotyp än personer med två mutationer och svarar bättre på järnbehandling (Donker et al., 2016). Intressant nog har flera TMPRSS6 SNPs visat sig ge känslighet för järnbrist i vissa populationer (An et al., 2012) och hos blodgivare (Sorensen et al., 2019).
En digenisk nedärvning har rapporterats hos en femårig kvinna som ursprungligen visade sig ha en atypisk IRIDA-genotyp med en TMPRSS6 (I212T) kausal och en (R271Q) tyst mutation (De Falco et al., 2014). Hon fick senare diagnosen Fibrodysplasia ossificans progressiva (FOP), en sällsynt dominant sjukdom med ektopisk benbildning i mjuka vävnader på grund av muterad BMP typ I-receptorgen ACVR1, som kodar för ALK2 (Shore et al., 2006). Den patologiska allelen ALK2R258S är konstitutivt aktiv eftersom mutationen påverkar genens glycin-serinrika domän och gör BMP/SMAD-banan överaktiv då den inte kan binda sin specifika hämmare FKBP12 (Pagani et al., 2017).
Detta sällsynta fall är särskilt illustrativt. För det första, eftersom ALK2:s glycin-serinrika domän interagerar med FKBP12 och mutationen destabiliserar bindningen, har det avslöjat en tidigare oväntad roll för FKBP12 som en regulator av ALK2 och hepcidin i levern (Colucci et al., 2017). För det andra har det lett till att identifiera en koppling mellan aktivering av ben- och lever BMP typ I-receptorer. För det tredje stärker fallet relevansen av intakt TMPRSS6 i kontrollen av den hepatiska BMP/SMAD-signalering, eftersom ingen IRIDA identifierades bland andra FOP-patienter med samma ACVR1-mutation och förmodligen normal TMPRSS6 (Pagani et al., 2017). Slutligen är detta fall förenligt med konceptet att TMPRSS6 haploinsufficiens inte kan orsaka klassisk IRIDA.
Den optimala behandlingen av IRIDA är odefinierad. Oralt järn är ineffektivt eftersom det inte absorberas. Tillägg av C-vitamin möjliggör sporadisk respons. Intravenöst järn inducerar ett partiellt svar vanligtvis i en långsammare takt jämfört med patienter med förvärvad järnbrist. EPO är ineffektivt i klassiska fall (De Falco et al., 2013; Heeney och Finberg, 2014).
Anemi till följd av hepcidinproducerande adenom
Detta är ett extremt sällsynt tillstånd hos vuxna patienter som är drabbade av glykogenlagringssjukdom 1a, en recessiv sjukdom på grund av brist på glukos-6-fosfatas, som katalyserar en reaktion som är involverad i både glykogenolysen och glukoneogenesen. Ett vanligt farligt sjukdomssymptom är hypoglykemi. Den nuvarande behandlingen leder till förlängd överlevnad hos drabbade barn upp till vuxen ålder med förekomst av flera komplikationer, t.ex. anemi och leveradenom. Anemin är mikrocytisk och hypokrom, järnbrist och refraktär mot oral järnbehandling. Anemin återkom efter kirurgisk resektion av adenom. Adenomvävnad visade sig vara positiv för hepcidin mRNA, medan normal omgivande vävnad uppvisade hepcidinundertryckning, vilket var förväntat på grund av den ektopiska okontrollerade hepcidinproduktionen (Weinstein et al., 2002). De hematologiska egenskaperna hos patienterna liknar dem hos IRIDA eftersom de delar höga hepcidinnivåer som en gemensam mekanism för anemi.
Inflammationsanemi
Inflammationsanemi (AI), tidigare känd som anemi vid kroniska sjukdomar, är en måttlig normokrom-normocytär anemi som utvecklas vid förhållanden med systemisk inflammation och immunaktivering. Den förekommer vid flera vanliga sjukdomar, inklusive kroniska infektioner, autoimmuna sjukdomar, avancerad cancer, kronisk njursjukdom, kongestiv hjärtsvikt, kronisk obstruktiv lungsjukdom, anemi hos äldre (åtminstone delvis) och graft versus host disease. AI är en av de vanligaste anemierna i världen och den vanligaste anemin hos sjukhusvårdade patienter. Akut inflammation bidrar till svårighetsgraden av anemi på intensivvårdsavdelningar. De molekylära mekanismer som ligger till grund för AI är många och komplexa. Överproduktion av cytokiner som IL1-β, TNF-α och IL-6 av makrofager och INF-γ av lymfocyter trubbar EPO-produktionen, försämrar erytropoesisresponsen, ökar hepcidinnivåerna och kan aktivera erytrofagocytos, särskilt i de akuta formerna (Weiss och Goodnough, 2005; Ganz, 2019).
Hepcidin aktiveras av IL-6 genom IL-6-receptorn (IL-6R) och JAK2-STAT3-signalering. Fullständig hepcidinaktivering kräver en aktiv BMP-SMAD-väg eftersom inaktivering av BMP-signalering minskar hepcidin i djurmodeller av inflammation (Theurl et al., 2011). Avregleringen av den systemiska järnhomeostasen orsakar järnsekvestrering av makrofager och minskad absorption och återvinning som leder till låg mättnad av transferrin och järnrestriktion av erytropoesin och andra vävnader.
Traditionell behandling av AI bygger på reversibilitet/kontroll av den underliggande sjukdomen, när det är möjligt. Om sjukdomen är obehandlingsbar och anemin är lindrig krävs en noggrann bedömning av risker-nytta för att undvika biverkningar av all behandling. Patofysiologibaserade behandlingar är begränsade till erytropoietinliknande föreningar och järn. Användningen av erytropoesistimulerande medel (ESA) undertrycker hepcidin genom att inducera erytropoesiexpansion. Detta tillvägagångssätt används i stor utsträckning hos patienter med kronisk njursjukdom, myelodysplastiska syndrom med låg risk och cancer som genomgår kemoterapi. En noggrann klinisk kontroll är dock nödvändig eftersom höga doser har kardiovaskulära biverkningar. Administrering av intravenöst järn kan lindra järnrestriktion som orsakas av ESA-beroende expansion av erytropoesin. Oralt järn är vanligtvis ineffektivt eftersom de höga hepcidinnivåerna motverkar dess tarmabsorption. Inhibitorer av prolylhydroxylas (hypoxiinducerbar faktor, HIF-stabilisatorer) är experimentella vid kronisk njursjukdom, i syfte att öka endogent EPO. Kronisk behandling med transfusioner av röda blodkroppar rekommenderas inte på grund av övergående effekt och biverkningar; den är begränsad till svår refraktär anemi (Camaschella, 2019; Weiss et al., 2019).
Anemier som är associerade med låga hepcidinnivåer
Ineffektiv erytropoes och låga eller olämpligt normala hepcidinnivåer med åtföljande järnöverbelastning är typiska kännetecken för de ”järnbelastande anemierna”.” Prototypen är β-thalassemi, en genetisk recessiv sjukdom som beror på mutationer i β-globingenen som orsakar anemi och överproduktion av α-globinkedjor. Den senare fälls ut som hemikromer i benmärgen, vilket skadar mognande erytroida prekursorer och leder till ineffektiv erytropoesi. Detta inträffar vid icke-transfusionsberoende thalassemi eller thalassemi intermedia, vars erytropoesi kännetecknas av förekomsten av omogna celler som frisätter erytroferron för att hämma leverhepcidinuttrycket. Hepcidinnivåerna är vanligtvis högre vid transfusionsberoende thalassemi, där den endogena ineffektiva erytropoesen åtminstone delvis undertrycks av transfusioner (Camaschella och Nai, 2016).
Hepcidinundertryckning medieras av det ökade cytokinet erytroferron (ERFE), en medlem av TNF-α-familjen som kodas av ERFE-genen, som syntetiseras av erytroblaster vid EPO-stimulering (Kautz et al., 2014). ERFE frigörs i cirkulationen och sekvenserar BMP:er, särskilt BMP6 (Arezes et al., 2018), vilket dämpar hepcidinsignalering som svar på järn. Dessutom sker ett epigenetiskt undertryckande vid hepcidinlocus av histondeacetylas HDAC3 (Pasricha et al., 2017). När anemi orsakar hypoxi, kommer andra mediatorer som PDGF-BB (Sonnweber et al, 2014), som frisätts av olika celltyper, undertrycker hepcidin.
Hepcidinnivåerna minskar genom en särskild mekanism vid lågriskmyelodysplasi med ringformade sideroblaster, en klonal sjukdom som beror på mutationer i spliceosomgenen SF3B1. Järn ackumuleras i mitokondrier, vilket leder till ineffektiv erytropoesi och systemisk järnöverbelastning. Ett onormalt spliced, förlängt ERFE-protein är mer kraftfullt än ERFE av vildtyp när det gäller att undertrycka hepcidin (Bondu et al, 2019) och orsakar transfusionsoberoende järnbelastning.
Targeted Therapies for Hepcidin-Related Anemias
Identifieringen av molekylära mekanismer som är ansvariga för de tidigare diskuterade anemierna har stimulerat forskningen om att utveckla riktade terapier för att ersätta nuvarande symtomatiska behandling (Sebastiani et al., 2016; Crielaard et al., 2017). Tillvägagångssätten skiljer sig åt beroende på typen av anemi och målet att minska eller öka hepcidinnivåerna eller deras effekter (tabell 2).
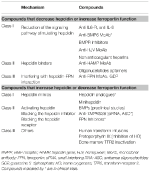
Tabell 2. Experimentella terapier med inriktning på hepcidin-ferroportin-axeln.
Experimentella terapier för att minska hepcidinnivåerna/öka ferroportin-funktionen
Med undantag för hepcidinproducerande tumörer, som måste avlägsnas kirurgiskt, kan föreningar som antagoniserar hepcidin eller dess effekter vara användbara vid alla anemier som kännetecknas av höga hepcidinnivåer. De skulle främst kunna användas vid kroniska inflammatoriska sjukdomar för att vända hypoferremia och anemi. Flera experimentella behandlingar som syftar till att manipulera hepcidinvägen och dess funktion har undersökts i prekliniska studier. Hepcidinantagonister är hämmare av hepcidinsyntesen/regulatorerna (Ganz, 2019), hepcidinbindare som blockerar dess funktion och föreningar som stör hepcidin-ferroportininteraktionen (tabell 2). Vissa föreningar är i klinisk prövning särskilt vid kronisk njursjukdom (Sheetz et al., 2019). I IRIDA har manipulation av hepcidinvägen föreslagits i prekliniska studier med användning av anti-HJV MoAb (Kovac et al., 2016).
Experimentella terapier för att öka hepcidinnivåerna/minskad ferroportinfunktion
Öka hepcidinnivåerna kan inte bara minska järnöverbelastningen utan också delvis kontrollera ineffektiv erytropoesi vid järnbelastningsanemier. β-thalassemi är det mest studerade av dessa tillstånd (Casu et al., 2018; Gupta et al., 2018). Föreslagna läkemedel är hepcidinanaloger (vissa i kliniska prövningar), hepcidinmodulatorer, särskilt TMPRSS6-hämmare, eller föreningar som interfererar med hepcidin-ferroportininteraktionen som minskar järnexporten (tabell 2).
Mediciner som ökar hepcidin minskar ineffektiv erytropoes på grund av den onda cirkeln mellan ineffektiv erytropoes och järnbelastning (Camaschella och Nai, 2016), medan läkemedel som gynnar mognad av erytroida prekursorer, som aktivinreceptor IIB-ligandfångaren, luspatercept, inte bara förbättrar anemi utan även förbättrar järnhomeostasen genom att minska hepcidinhämningen (Piga et al, 2019).
Vissa riktade tillvägagångssätt som nu är föremål för kliniska prövningar kommer förhoppningsvis att resultera i nya behandlingar för en rad olika anemier.
Slutsats
De spektakulära framstegen i förståelsen av regleringen av järnmetabolismen och hepcidin möjliggjorde en bättre förståelse för erytropoesikontrollen, eftersom järn tillsammans med erytropoetin är en grundläggande faktor för erytroida cellers mognad. Tillstånd som leder till anemi kan förknippas med höga och låga hepcidinnivåer. I båda fallen kan kontrasterande hepcidinavreglering förbättra/korrigera anemi i prekliniska modeller, vilket erbjuder nya verktyg som redan är eller snart kommer att bli kliniskt utforskade för behandling av specifika anemier.
Författarbidrag
AP utarbetade artikeln. CC utarbetade den slutliga versionen. AN och LS bidrog till att skriva och kritiskt granska manuskriptet. Alla författare godkände den slutliga versionen.
Finansiering
Denna artikel stöddes delvis av ett EHA Advanced Research Grant 2018 till AP. Cariplo Foundation Young Investigator Grant n° 2017-0916 till AN.
Intressekonflikt
CC är konsult för Vifor Pharma, Celgene och Novartis.
Resten av författarna förklarar att forskningen utfördes i avsaknad av kommersiella eller finansiella relationer som skulle kunna uppfattas som en potentiell intressekonflikt.
An, P., Wu, Q., Wang, H., Guan, Y., Mu, M., Liao, Y., et al. (2012). TMPRSS6-, men inte TF-, TFR2- eller BMP2-varianter är associerade med ökad risk för järnbristanemi. Hum. Mol. Genet. 21, 2124-2131. doi: 10.1093/hmg/dds028
PubMed Abstract | CrossRef Full Text | Google Scholar
Andriopoulos, B. Jr., Corradini, E., Xia, Y., Faasse, S. A., Chen, S., Grgurevic, L., et al. (2009). BMP6 är en viktig endogen regulator av hepcidinuttryck och järnmetabolism. Nat. Genet. 41, 482-487. doi: 10.1038/ng.335
PubMed Abstract | CrossRef Full Text | Google Scholar
Arezes, J., Foy, N., McHugh, K., Sawant, A., Quinkert, D., Terraube, V., et al. (2018). Erytroferron hämmar induktionen av hepcidin av BMP6. Blood 132, 1473-1477. doi: 10.1182/blood-2018-06-857995
PubMed Abstract | CrossRef Full Text | Google Scholar
Aschemeyer, S., Qiao, B., Stefanova, D., Valore, E. V., Sek, A. C., Ruwe, T. A., et al. (2018). Struktur-funktionsanalys av ferroportin definierar bindningsstället och en alternativ verkningsmekanism för hepcidin. Blood. 131, 899-910. doi: 10.1182/blood-2017-05-786590
CrossRef Full Text | Google Scholar
Babitt, J. L., Huang, F. W., Wrighting, D. M., Xia, Y., Sidis, Y., Samad, T. A., et al. (2006). Signalering av benmorfogenetiskt protein genom hemojuvelin reglerar hepcidinuttrycket. Nat. Genet. 38, 531-539. doi: 10.1038/ng1777
PubMed Abstract | CrossRef Full Text | Google Scholar
Beguin, Y. (2003). Löslig transferrinreceptor för utvärdering av erytropoesi och järnstatus. Clin. Chim. Acta 329, 9-22. doi: 10.1016/S0009-8981(03)00005-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Bondu, S., Alary, A. S., Lefevre, C., Houy, A., Jung, G., Lefebvre, T., et al. (2019). En variant erythroferron stör järnhomeostasen vid SF3B1-muterat myelodysplastiskt syndrom. Sci. Transl. Med. 11:pii:eaav5467. doi: 10.1126/scitranslmed.aav5467
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C. (2013). Hur jag hanterar patienter med atypisk mikrocytisk anemi. Br. J. Haematol. 160, 12-24. doi: 10.1111/bjh.12081
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C. (2019). Järnbrist. Blood 133, 30-39. doi: 10.1182/blood-2018-05-81515944
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C., and Nai, A. (2016). Ineffektiv erytropoes och reglering av järnstatus vid järnbelastningsanemi. Br. J. Haematol. 172, 512-523. doi: 10.1111/bjh.13820
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C. och Pagani, A. (2018). Framsteg i förståelsen av järnmetabolism och dess crosstalk med erytropoiesis. Br. J. Haematol. 182, 481-494. doi: 10.1111/bjh.15403
PubMed Abstract | CrossRef Full Text | Google Scholar
Canali, S., Wang, C. Y., Zumbrennen-Bullough, K. B., Bayer, A. och Babitt, J. L. (2017). Bone morphogenetic protein 2 kontrollerar järnhomeostas hos möss oberoende av Bmp6. Am. J. Hematol. 92, 1204-1213. doi: 10.1002/ajh.24888
PubMed Abstract | CrossRef Full Text | Google Scholar
Casu, C., Nemeth, E., and Rivella, S. (2018). Hepcidinagonister som terapeutiska verktyg. Blood 131, 1790-1794. doi: 10.1182/blood-2017-11-737411
PubMed Abstract | CrossRef Full Text | Google Scholar
Colucci, S., Pagani, A., Pettinato, M., Artuso, I., Nai, A., Camaschella, C., et al. (2017). Immunofilin FKBP12 hämmar hepcidinuttryck genom att binda BMP typ I-receptorn ALK2 i hepatocyter. Blood 130, 2111-2120. doi: 10.1182/blood-2017-04-780692
PubMed Abstract | CrossRef Full Text | Google Scholar
Crielaard, B. J., Lammers, T. och Rivella, S. (2017). Att rikta in sig på järnmetabolism vid upptäckt och leverans av läkemedel. Nat. Rev. Drug Discov. 16, 400-423. doi: 10.1038/nrd.2016.248
PubMed Abstract | CrossRef Full Text | Google Scholar
De Falco, L., Sanchez, M., Silvestri, L., Kannengiesser, C., Muckenthaler, M. U., Iolascon, A., et al. (2013). Järnrefraktär järnbristanemi. Haematologica 98, 845-853. doi: 10.3324/haematol.2012.075515
PubMed Abstract | CrossRef Full Text | Google Scholar
De Falco, L., Silvestri, L., Kannengiesser, C., Moran, E., Oudin, C., Rausa, M., et al. (2014). Funktionell och klinisk inverkan av nya TMPRSS6-varianter hos patienter med järnrefraktär järnbristanemi och genotyp-fenotypstudier. Hum. Mutat. 35, 1321-1329. doi: 10.1002/humu.22632
PubMed Abstract | CrossRef Full Text | Google Scholar
Donker, A. E., Schaap, C. C., Novotny, V. M., Smeets, R., Peters, T. M., van den Heuvel, B. L., et al. (2016). Järnrefraktär järnbristanemi: en heterogen sjukdom som inte alltid är järnrefraktär. Am. J. Hematol. 91, E482-E490. doi: 10.1002/ajh.24561
PubMed Abstract | CrossRef Full Text | Google Scholar
Du, X., She, E., Gelbart, T., Truksa, J., Lee, P., Xia, Y., et al. (2008). Serinproteaset TMPRSS6 krävs för att känna av järnbrist. Science 320, 1088-1092. doi: 10.1126/science.1157121
PubMed Abstract | CrossRef Full Text | Google Scholar
Finberg, K. E., Heeney, M. M., Campagna, D. R., Aydinok, Y., Pearson, H. A., Hartman, K. R., et al. (2008). Mutationer i TMPRSS6 orsakar järnrefraktär järnbristanemi (IRIDA). Nat. Genet. 40, 569-571. doi: 10.1038/ng.130
PubMed Abstract | CrossRef Full Text | Google Scholar
Ganz, T. (2019). Anemi till följd av inflammation. N. Engl. J. Med. 381, 1148-1157. doi: 10.1056/NEJMra1804281
PubMed Abstract | CrossRef Full Text | Google Scholar
Gupta, R., Musallam, K. M., Taher, A. T. och Rivella, S. (2018). Ineffektiv erytropoesi: anemi och järnöverbelastning. Hematol. Oncol. Clin. North Am. 32, 213-221. doi: 10.1016/j.hoc.2017.11.009
PubMed Abstract | CrossRef Full Text | Google Scholar
Heeney, M. M., and Finberg, K. E. (2014). Järnrefraktär järnbristanemi (IRIDA). Hematol. Oncol. Clin. North Am. 28, 637-652. doi: 10.1016/j.hoc.2014.04.009
PubMed Abstract | CrossRef Full Text | Google Scholar
Heeney, M. M., Guo, D., De Falco, L., Campagna, D. R., Olbina, G., Kao, P. P., et al. (2018). Normaliserande hepcidin förutsäger TMPRSS6-mutationsstatus hos patienter med kronisk järnbrist. Blood 132, 448-452. doi: 10.1182/blood-2017-03-773028
PubMed Abstract | CrossRef Full Text | Google Scholar
Kautz, L., Jung, G., Valore, E. V., Rivella, S., Nemeth, E. och Ganz, T. (2014). Identifiering av erytroferron som en erytroid regulator av järnmetabolismen. Nat. Genet. 46, 678-684. doi: 10.1038/ng.2996
PubMed Abstract | CrossRef Full Text | Google Scholar
Koch, P. S., Olsavszky, V., Ulbrich, F., Sticht, C., Demory, A., Leibing, T., et al. (2017). Angiokrin Bmp2-signalering i murinlever kontrollerar normal järnhomeostas. Blood 129, 415-419. doi: 10.1182/blood-2016-07-729822
PubMed Abstract | CrossRef Full Text | Google Scholar
Kovac, S., Boser, P., Cui, Y., Ferring-Appel, D., Casarrubea, D., Huang, L., et al. (2016). Anti-hemojuvelin-antikropp korrigerar anemi orsakad av olämpligt höga hepcidinnivåer. Haematologica 101, e173-e176. doi: 10.3324/haematol.2015.140772
PubMed Abstract | CrossRef Full Text | Google Scholar
Lakhal, S., Schodel, J., Townsend, A. R., Pugh, C. W., Ratcliffe, P. J. och Mole, D. R. (2011). Reglering av typ II transmembran serinproteinas TMPRSS6 av hypoxiinducerbara faktorer: ny länk mellan hypoxi-signalering och järnhomeostas. J. Biol. Chem. 286, 4090-4097. doi: 10.1074/jbc.M110.173096
PubMed Abstract | CrossRef Full Text | Google Scholar
Meynard, D., Kautz, L., Darnaud, V., Canonne-Hergaux, F., Coppin, H. och Roth, M. P. (2009). Brist på det benmorfogenetiska proteinet BMP6 inducerar massiv järnöverbelastning. Nat. Genet. 41, 478-481. doi: 10.1038/ng.320
PubMed Abstract | CrossRef Full Text | Google Scholar
Muckenthaler, M. U., Rivella, S., Hentze, M. W., and Galy, B. (2017). En röd matta för järnmetabolism. Cell 168, 344-361. doi: 10.1016/j.cell.2016.12.034
PubMed Abstract | CrossRef Full Text | Google Scholar
Nemeth, E., Tuttle, M. S., Powelson, J., Vaughn, M. B., Donovan, A., Ward, D. M., et al. (2004). Hepcidin reglerar cellulär järnutflöde genom att binda till ferroportin och inducera dess internalisering. Science 306, 2090-2093. doi: 10.1126/science.1104742
PubMed Abstract | CrossRef Full Text | Google Scholar
Pagani, A., Colucci, S., Bocciardi, R., Bertamino, M., Dufour, C., Ravazzolo, R., et al. (2017). En ny form av IRIDA på grund av kombinerade heterozygota mutationer av TMPRSS6 och ACVR1A som kodar för BMP-receptorn ALK2. Blood 129, 3392-3395. doi: 10.1182/blood-2017-03-773481
PubMed Abstract | CrossRef Full Text | Google Scholar
Pagani, A., Vieillevoye, M., Nai, A., Rausa, M., Ladli, M., Lacombe, C., et al. (2015). Reglering av transferrinreceptor-2 på cellytan genom järnberoende klyvning och frisättning av en löslig form. Haematologica 100, 458-465. doi: 10.3324/haematol.2014.118521
PubMed Abstract | CrossRef Full Text | Google Scholar
Pasricha, S. R., Lim, P. J., Duarte, T. L., Casu, C., Oosterhuis, D., Mleczko-Sanecka, K., et al. (2017). Hepcidin regleras av promotorassocierad histonacetylering och HDAC3. Nat. Commun. 8:403. doi: 10.1038/s41467-017-00500-z
PubMed Abstract | CrossRef Full Text | Google Scholar
Piga, A., Perrotta, S., Gamberini, M. R., Voskaridou, E., Melpignano, A., Filosa, A., et al. (2019). Luspatercept förbättrar hemoglobinnivåer och behov av blodtransfusioner i en studie av patienter med beta-thalassemi. Blood 133, 1279-1289. doi: 10.1182/blood-2018-10-879247
PubMed Abstract | CrossRef Full Text | Google Scholar
Rausa, M., Pagani, A., Nai, A., Campanella, A., Gilberti, M. E., Apostoli, P., et al. (2015). Bmp6-uttryck i icke parenkymala celler i murinlever: en mekanism för att kontrollera deras höga järnexportörsaktivitet och skydda hepatocyter från järnöverbelastning? PLoS One 10:e0122696. doi: 10.1371/journal.pone.0122696
PubMed Abstract | CrossRef Full Text | Google Scholar
Sebastiani, G., Wilkinson, N. och Pantopoulos, K. (2016). Farmakologisk inriktning av hepcidin/ferroportin-axeln. Front. Pharmacol. 7:160. doi: 10.3389/fphar.2016.00160
PubMed Abstract | CrossRef Full Text | Google Scholar
Sheetz, M., Barrington, P., Callies, S., Berg, P. H., McColm, J., Marbury, T., et al. (2019). Att rikta in sig på hepcidin-ferroportinvägen vid anemi vid kronisk njursjukdom. Br. J. Clin. Pharmacol. 85, 935-948. doi: 10.1111/bcp.13877
PubMed Abstract | CrossRef Full Text | Google Scholar
Shore, E. M., Xu, M., Feldman, G. J., Fenstermacher, D. A., Cho, T. J., Choi, I. H., et al. (2006). En återkommande mutation i BMP typ I-receptorn ACVR1 orsakar ärftlig och sporadisk fibrodysplasia ossificans progressiva. Nat. Genet. 38, 525-527. doi: 10.1038/ng1783
PubMed Abstract | CrossRef Full Text | Google Scholar
Silvestri, L., Pagani, A., Nai, A., De Domenico, I., Kaplan, J. och Camaschella, C. (2008). Serinproteaset matriptas-2 (TMPRSS6) hämmar hepcidinaktivering genom att klyva membranhemojuvelin. Cell Metab. 8, 502-511. doi: 10.1016/j.cmet.2008.09.012
PubMed Abstract | CrossRef Full Text | Google Scholar
Silvestri, L., Rausa, M., Pagani, A., Nai, A. och Camaschella, C. (2013). Hur bedömer man kausaliteten hos TMPRSS6-mutationer? Hum. Mutat. 34, 1043-1045. doi: 10.1002/humu.22321
PubMed Abstract | CrossRef Full Text | Google Scholar
Sonnweber, T., Nachbaur, D., Schroll, A., Nairz, M., Seifert, M., Demetz, E., et al. (2014). Hypoxiinducerad nedreglering av hepcidin medieras av platelet derived growth factor BB. Gut 63, 1951-1959. doi: 10.1136/gutjnl-2013-305317
PubMed Abstract | CrossRef Full Text | Google Scholar
Sorensen, E., Rigas, A. S., Didriksen, M., Burgdorf, K. S., Thorner, L. W., Pedersen, O. B., et al. (2019). Genetiska faktorer som påverkar hemoglobinnivåerna hos 15 567 blodgivare: resultat från den danska blodgivarundersökningen. Transfusion 59, 226-231. doi: 10.1111/trf.15075
CrossRef Full Text | Google Scholar
Theurl, I., Schroll, A., Sonnweber, T., Nairz, M., Theurl, M., Willenbacher, W., et al. (2011). Farmakologisk hämning av hepcidinuttryck reverserar anemi av kronisk inflammation hos råttor. Blood 118, 4977-4984. doi: 10.1182/blood-2011-03-345066
PubMed Abstract | CrossRef Full Text | Google Scholar
Weinstein, D. A., Roy, C. N., Fleming, M. D., Loda, M. F., Wolfsdorf, J. I. och Andrews, N. C. (2002). Olämpligt uttryck av hepcidin är förknippat med järnrefraktär anemi: konsekvenser för anemi vid kroniska sjukdomar. Blood 100, 3776-3781. doi: 10.1182/blood-2002-04-1260
PubMed Abstract | CrossRef Full Text | Google Scholar
Weiss, G., Ganz, T., and Goodnough, L. T. (2019). Anemi till följd av inflammation. Blood 133, 40-50. doi: 10.1182/blood-2018-06-856500
PubMed Abstract | CrossRef Full Text | Google Scholar
Weiss, G., and Goodnough, L. T. (2005). Anemi vid kronisk sjukdom. N. Engl. J. Med. 352, 1011-1023. doi: 10.1056/NEJMra041809
PubMed Abstract | CrossRef Full Text | Google Scholar
.
Leave a Reply