Frontiers in Physiology
Introduction
Anämie ist eine der häufigsten Erkrankungen weltweit, und Anämie aufgrund von Eisenmangel ist nach mehreren Analysen die vorherrschende Form (Übersicht in Camaschella, 2019). Diese Art von Anämie resultiert aus dem Gesamtkörpereisenmangel und der Unfähigkeit, die große Menge an Eisen zu liefern, die das Knochenmark verbraucht, um eine ausreichende Anzahl roter Blutkörperchen zu produzieren, um die Sauerstoffversorgung des Gewebes aufrechtzuerhalten.
Die Eisenverfügbarkeit wird durch das Leberpeptidhormon Hepcidin gesteuert. Der Anstieg des Körpereisens bewirkt die Produktion von Hepcidin, das in den Blutkreislauf freigesetzt wird und auf seinen Rezeptor Ferroportin wirkt, ein Transmembran-Eisenexportprotein, das auf Enterozyten, Makrophagen und Hepatozyten stark exprimiert wird. Hepcidin reduziert den Eiseneintrag in das Plasma aus absorbierenden Duodenalzellen und eisenrecycelnden Makrophagen, indem es den Eisenexport blockiert (Aschemeyer et al., 2018) und den Eisenexporteur Ferroportin abbaut (Nemeth et al., 2004). Durch die Regulierung des Plasmaeisens und der systemischen Eisenhomöostase hat die Hepcidin/Ferroportin-Achse einen starken Einfluss auf die Erythropoese und damit auf die mögliche Entwicklung einer Anämie.
Der Zusammenhang zwischen Eisen und Erythropoese
Der Prozess der Produktion roter Blutkörperchen verbraucht etwa 80 % des zirkulierenden Eisens für die Hämoglobinsynthese der reifenden Erythroblasten. Der größte Teil des Eisens (20-25 mg/Tag) wird von Makrophagen recycelt, während eine begrenzte Menge (1-2 mg/Tag) aus der intestinalen Absorption stammt. Das Nierenhormon Erythropoetin (EPO) steuert die Proliferation der erythroiden Vorläuferzellen, insbesondere der CFU-e und in geringerem Maße der BFU-e, sowie die frühe Phase der terminalen Erythropoese, während der Eisenbedarf in den späten Differenzierungsstadien von den Proerythroblasten zu den Retikulozyten für die Synthese des in das Hämoglobin eingebauten Häms erhöht ist (Muckenthaler et al, 2017).
Die Regulierung von Hepcidin erfordert einen Crosstalk zwischen leberendothelialen sinusoidalen Zellen (LSEC), die Knochenmorphogenetische Proteine (BMPs) zur Aktivierung des BMP-SMAD-Signalwegs produzieren, und Hepatozyten, die Hepcidin produzieren und freisetzen (Babitt et al., 2006; Rausa et al., 2015). BMP6 und BMP2 sind die wichtigsten BMPs, die Hepcidin hochregulieren, während die BMP6-Expression eisenabhängig ist (Andriopoulos et al., 2009; Meynard et al., 2009), scheint BMP2 weniger eisenabhängig zu sein (Canali et al., 2017; Koch et al., 2017).
Die Hepcidinspiegel sind bei absolutem Eisenmangel und Eisenmangelanämie niedrig. Unter diesen Bedingungen sind die Eisenspeicher erschöpft und die BMP-SMAD-Signalgebung wird auf mehreren Ebenen ausgeschaltet. Erstens wird die BMP6-Expression unterdrückt, zweitens ist die Aktivität von TMPRSS6, einer Protease, die den BMP-Co-Rezeptor Hemojuvelin spaltet (Silvestri et al., 2008), stark erhöht (Lakhal et al., 2011), und drittens unterdrückt Histon-Deacetylase3 (HDAC3) den Hepcidin-Locus (Pasricha et al., 2017). Bei Eisenmangel ist die Verringerung der Hepcidin-Produktion ein Anpassungsmechanismus, der die diätetische und pharmakologische Eisenabsorption erleichtert (Camaschella und Pagani, 2018).
Bei schwerer Anämie stimuliert die gleichzeitig bestehende Hypoxie die Erythropoese durch eine erhöhte Nierensynthese und Freisetzung von EPO. Dies führt zu einer Unterdrückung der Hepcidin-Transkription durch Erythroferron (ERFE), ein EPO-Zielgen, das von Erythroblasten produziert wird (Kautz et al., 2014), durch Moleküle (z. B. PDGF-BB), die von anderen Geweben freigesetzt werden (Sonnweber et al., 2014), und wahrscheinlich durch lösliche Komponenten von Transferrinrezeptoren (TFR), sTFR1 (Beguin, 2003) und sTFR2 (Pagani et al., 2015). Das endgültige Ziel ist es, genügend Eisen für den Bedarf einer erweiterten Erythropoese bereitzustellen.
Anämien mit abnormalen Hepcidinspiegeln
Anämien können auf der Grundlage der Hepcidinspiegel als Anämien mit hohem und niedrigem Hepcidin klassifiziert werden. Es ist intuitiv, dass anhaltend hohe Hepcidinspiegel durch die Blockierung der Eisenabsorption eine Eisenmangelanämie verursachen, da die Erythropoese mit weniger Eisen versorgt wird. Umgekehrt ist eine ineffektive Erythropoese kennzeichnend für die so genannten eisenlastigen Anämien mit niedrigen Hepcidinspiegeln und Eisenüberladung. Diese beiden Gruppen von Anämien sind das Ergebnis entgegengesetzter pathophysiologischer Mechanismen (Abbildung 1). In der ersten Gruppe ist die Anämie auf die hemmende Wirkung von Hepcidin auf die Eisenabsorption und das Eisenrecycling zurückzuführen, was zu einem systemischen Eisenmangel führt; in der zweiten Gruppe ist die Anämie auf die Unterdrückung von Hepcidin durch eine erweiterte abnorme Erythropoese zurückzuführen (Camaschella und Nai, 2016).
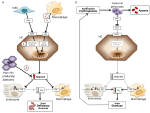
Abbildung 1. Schematische Darstellung der Mechanismen von Anämien mit hohem (linke Tafel) und niedrigem Hepcidin (rechte Tafel). Tafel (A). Molekulare Pathogenese der mit hohen Hepcidinwerten assoziierten Anämie. LSEC, Lebersinusoidale Endothelzellen, die knochenmorphogenetische Proteine (BMPs) produzieren; BMPRs, BMP-Rezeptoren; IL6, Interleukin 6; HC, Hepatozyten; HAMP, Hepcidin-Gen. Fe, Eisen; FPN, Ferroportin; 1, IRIDA; 2, Entzündungsanämie; 3, Hepcidin produzierendes Adenom. Tafel (B). Molekulare Pathogenese der Hepcidin-Variation bei Anämien, die auf eine ineffektive Erythropoese zurückzuführen sind. ERFE, Erythroferron sequestrierende BMPs. Andere Mechanismen, die Hepcidin bei dieser Art von Anämie hemmen, wie die Abnahme der Transferrinsättigung und Hypoxie, sind nicht dargestellt. Einzelheiten siehe Text.
Anämie im Zusammenhang mit hohen Hepcidinspiegeln
Diese Gruppe umfasst zwei vererbte seltene Störungen (eisenrefraktäre Eisenmangelanämie und Hepcidin-produzierende Adenome bei einem angeborenen Fehler des Glukosestoffwechsels) und eine erworbene häufige Erkrankung: Anämie der Entzündung (Tabelle 1).
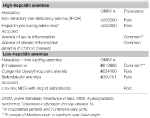
Tabelle 1. Anämien, klassifiziert nach Hepcidinspiegeln.
Eisenrefraktäre Eisenmangelanämie
Die eisenrefraktäre Eisenmangelanämie (IRIDA) ist eine seltene rezessive Störung, die durch hypochrome mikrozytäre Anämie, niedrige Transferrinsättigung und unangemessen normale/hohe Hepcidinspiegel gekennzeichnet ist. Sie wird durch Mutationen von TMPRSS6 (Finberg et al., 2008) verursacht, einem Gen, das für die Serinprotease vom Typ II, Matriptase-2 (Du et al., 2008), kodiert. Mutationen von TMPRSS6 sind entlang des Gens verteilt und können verschiedene Domänen betreffen, insbesondere die katalytische Domäne (De Falco et al., 2014). Diese Transmembranprotease, die in der Leber in hohem Maße exprimiert wird, hemmt die Hepcidin-Transkription, indem sie den Zelloberflächen-BMP-Korezeptor Hemojuvelin spaltet und so die BMP-Signalübertragung und die Hepcidin-Synthese abschwächt (Silvestri et al., 2008). Die Funktion von TMPRSS6 ist bei Eisenmangel unerlässlich, um den Kompensationsmechanismus einer erhöhten Eisenaufnahme zu ermöglichen.
IRIDA ist von Geburt an vorhanden und wird in der Regel in der Kindheit diagnostiziert. Im Vergleich zum klassischen Eisenmangel sind die Eisenparameter atypisch und lassen den Verdacht auf die Krankheit aufkommen. Die prozentuale Sättigung von Transferrin ist wie bei anderen Formen des Eisenmangels stark reduziert (weniger als 10 %); im Gegensatz zum Eisenmangel sind die Serumferritinwerte jedoch normal/erhöht (Camaschella, 2013; De Falco et al., 2013). Dies spiegelt eine erhöhte Ferritinakkumulation in Makrophagen wider, die auf hohe Hepcidinspiegel zurückzuführen ist, die eine Sequestrierung von Speichereisen induzieren.
Keiner der für die IRIDA-Diagnose vorgeschlagenen Tests deckt 100 % der Fälle ab. Der Gentest identifiziert TMPRSS6-Mutationen, die in einigen Fällen (Non-Sense-, Frame-Shift- und Spleißmutationen) eindeutig kausal sind. In anderen Fällen, wie z. B. bei bisher nicht gemeldeten Missense-Mutationen, sind funktionelle Studien erforderlich, um die Kausalität nachzuweisen (Silvestri et al., 2013). Diese Tests sind jedoch kaum verfügbar. Die Hepcidinwerte im Serum sind in der Regel unabhängig von Eisenmangel erhöht/normal und stimmen mit einem hohen/normalen Ferritinwert überein. Es ist wichtig, eine Entzündung durch die gleichzeitige Gabe von C-reaktivem Protein auszuschließen.
Es wurde berichtet, dass einige Patienten mit dem Phänotyp des refraktären Eisenmangels ein einzelnes mutiertes TMPRSS6-Allel aufweisen; hier wird diskutiert, ob sie als IRIDA angesehen werden sollten oder nicht. Man kann sich ein Spektrum von Zuständen vorstellen, das von klassischer schwerer IRIDA aufgrund homozygoter oder compound heterozygoter TMPRSS6-Mutationen bis hin zu erhöhter Anfälligkeit für Eisenmangel durch einzelne Mutationen/polymorphe Veränderungen reicht. Ein Ansatz, der zur Vorhersage von klassischem IRIDA vorgeschlagen wurde, ist die Normalisierung von Hepcidin auf andere Eisenparameter, wie das Verhältnis von Transferrinsättigung (Tsat)/log Hepcidin oder Tsat/log Ferritin (Donker et al., 2016). Anderen Autoren zufolge haben die meisten Patienten mit einem schweren IRIDA-Phänotyp biallelische TMPRSS6-Mutationen, und wenn sie nicht identifiziert werden, kann das zweite Allel genetisch verdeckt sein (Heeney et al., 2018). Im Allgemeinen haben Personen mit einem einzigen Allel einen milderen Phänotyp als Personen mit zwei Mutationen und sprechen besser auf eine Eisenbehandlung an (Donker et al., 2016). Interessanterweise wurde gezeigt, dass mehrere TMPRSS6-SNPs in einigen Populationen (An et al., 2012) und bei Blutspendern (Sorensen et al., 2019) für Eisenmangel anfällig sind.
Ein digenischer Erbgang wurde bei einem fünfjährigen Mädchen festgestellt, das ursprünglich einen atypischen IRIDA-Genotyp mit einer kausalen TMPRSS6- (I212T) und einer stillen Mutation (R271Q) aufwies (De Falco et al., 2014). Später wurde bei ihr Fibrodysplasia ossificans progressiva (FOP) diagnostiziert, eine seltene dominante Erkrankung mit ektopischer Knochenbildung im Weichteilgewebe, die auf ein mutiertes BMP-Typ-I-Rezeptor-Gen ACVR1 zurückzuführen ist, das für ALK2 kodiert (Shore et al., 2006). Das pathologische Allel ALK2R258S ist konstitutiv aktiv, da die Mutation die glycin-serinreiche Domäne des Gens betrifft und den BMP/SMAD-Signalweg überaktiv macht, da er nicht in der Lage ist, seinen spezifischen Inhibitor FKBP12 zu binden (Pagani et al., 2017).
Dieser seltene Fall ist besonders illustrativ. Erstens, da die ALK2-Glycin-Serin-reiche Domäne mit FKBP12 interagiert und die Mutation die Bindung destabilisiert, hat sie eine zuvor nicht vermutete Rolle für FKBP12 als Modulator von ALK2 und Hepcidin in der Leber aufgedeckt (Colucci et al., 2017). Zweitens hat es dazu geführt, eine Verbindung zwischen der Aktivierung von Knochen- und Leber-BMP-Typ-I-Rezeptoren zu identifizieren. Drittens stärkt der Fall die Relevanz von intaktem TMPRSS6 bei der Kontrolle der hepatischen BMP/SMAD-Signalgebung, da bei anderen FOP-Patienten mit der gleichen ACVR1-Mutation und vermutlich normalem TMPRSS6 kein IRIDA identifiziert wurde (Pagani et al., 2017). Schließlich steht dieser Fall im Einklang mit dem Konzept, dass TMPRSS6-Haploinsuffizienz keine klassische IRIDA verursachen kann.
Die optimale Behandlung von IRIDA ist nicht definiert. Orales Eisen ist unwirksam, da es nicht absorbiert wird. Der Zusatz von Vitamin C ermöglicht ein sporadisches Ansprechen. Intravenöses Eisen führt zu einem teilweisen Ansprechen, das im Vergleich zu Patienten mit erworbenem Eisenmangel in der Regel langsamer ist. EPO ist in klassischen Fällen unwirksam (De Falco et al., 2013; Heeney und Finberg, 2014).
Anämie der Hepcidin-produzierenden Adenome
Dies ist ein extrem seltener Zustand bei erwachsenen Patienten, die von der Glykogenspeicherkrankheit 1a betroffen sind, einer rezessiven Störung aufgrund eines Mangels an Glucose-6-Phosphatase, die eine Reaktion katalysiert, die sowohl an der Glykogenolyse als auch an der Glukoneogenese beteiligt ist. Ein häufiges gefährliches Krankheitssymptom ist Hypoglykämie. Die derzeitige Behandlung führt zu einem verlängerten Überleben der betroffenen Kinder bis zum Erwachsenenalter, wobei verschiedene Komplikationen wie Anämie und Leberadenome auftreten können. Die Anämie ist mikrozytär und hypochrom, weist einen Eisenmangel auf und ist refraktär gegenüber einer oralen Eisenbehandlung. Die Anämie bildete sich nach der chirurgischen Adenomresektion zurück. Das Adenomgewebe war positiv für Hepcidin-mRNA, während das umgebende normale Gewebe eine Hepcidin-Suppression aufwies, wie aufgrund der ektopischen unkontrollierten Hepcidin-Produktion zu erwarten war (Weinstein et al., 2002). Die hämatologischen Merkmale der Patienten ähneln denen von IRIDA, da sie hohe Hepcidinspiegel als gemeinsamen Mechanismus der Anämie aufweisen.
Entzündungsanämie
Entzündungsanämie (AI), früher bekannt als Anämie bei chronischen Krankheiten, ist eine moderate normochrom-normozytäre Anämie, die sich unter Bedingungen systemischer Entzündung und Immunaktivierung entwickelt. Sie tritt bei mehreren häufigen Erkrankungen auf, darunter chronische Infektionen, Autoimmunerkrankungen, Krebs im fortgeschrittenen Stadium, chronische Nierenerkrankungen, kongestive Herzinsuffizienz, chronisch obstruktive Lungenerkrankungen, Anämie bei älteren Menschen (zumindest teilweise) und Graft-versus-Host-Krankheit. Die AI ist eine der häufigsten Anämien weltweit und die häufigste Anämie bei hospitalisierten Patienten. Akute Entzündungen tragen zur Schwere der Anämie auf der Intensivstation bei. Die der AI zugrunde liegenden molekularen Mechanismen sind vielfältig und komplex. Die Überproduktion von Zytokinen wie IL1-β, TNF-α und IL-6 durch Makrophagen und INF-γ durch Lymphozyten hemmt die EPO-Produktion, beeinträchtigt die Erythropoese-Reaktion, erhöht den Hepcidinspiegel und kann die Erythrophagozytose aktivieren, insbesondere bei akuten Formen (Weiss und Goodnough, 2005; Ganz, 2019).
Hepcidin wird durch IL-6 über den IL-6-Rezeptor (IL-6R) und die JAK2-STAT3-Signalisierung aktiviert. Für eine vollständige Hepcidin-Aktivierung ist ein aktiver BMP-SMAD-Signalweg erforderlich, da die Inaktivierung der BMP-Signalisierung in Tiermodellen für Entzündungen zu einer Abnahme von Hepcidin führt (Theurl et al., 2011). Die Deregulierung der systemischen Eisenhomöostase führt zu einer Sequestrierung von Eisen durch Makrophagen und zu einer verminderten Absorption und Wiederverwertung, was zu einer niedrigen Sättigung von Transferrin und einer Eisenrestriktion in der Erythropoese und anderen Geweben führt.
Die traditionelle Behandlung von AI basiert auf der Reversibilität/Kontrolle der zugrunde liegenden Krankheit, wann immer dies möglich ist. Ist die Krankheit nicht behandelbar und die Anämie mild, ist eine sorgfältige Nutzen-Risiko-Abwägung erforderlich, um Nebenwirkungen jeder Behandlung zu vermeiden. Pathophysiologisch begründete Behandlungen beschränken sich auf Erythropoietin-ähnliche Verbindungen und Eisen. Die Verwendung von Erythropoese-stimulierenden Mitteln (ESA) unterdrückt Hepcidin, indem sie eine Ausweitung der Erythropoese induziert. Dieser Ansatz wird häufig bei Patienten mit chronischen Nierenerkrankungen, myelodysplastischen Syndromen mit niedrigem Risiko und Krebserkrankungen, die sich einer Chemotherapie unterziehen, eingesetzt. Allerdings ist eine sorgfältige klinische Kontrolle erforderlich, da hohe Dosen kardiovaskuläre Nebenwirkungen haben. Die Verabreichung von intravenösem Eisen kann die durch die ESA-abhängige Expansion der Erythropoese verursachte Eisenrestriktion beheben. Die orale Gabe von Eisen ist in der Regel unwirksam, da die hohen Hepcidinspiegel der intestinalen Absorption entgegenwirken. Inhibitoren der Prolylhydroxylase (hypoxia inducible factor, HIF-Stabilisatoren) werden bei chronischen Nierenerkrankungen experimentell eingesetzt, um das endogene EPO zu erhöhen. Eine chronische Behandlung mit Erythrozytentransfusionen wird wegen der vorübergehenden Wirkung und der Nebenwirkungen nicht empfohlen; sie ist auf schwere refraktäre Anämien beschränkt (Camaschella, 2019; Weiss et al., 2019).
Anämien, die mit niedrigen Hepcidinspiegeln assoziiert sind
Eine unwirksame Erythropoese und niedrige oder unangemessen normale Hepcidinspiegel mit daraus resultierender Eisenüberladung sind typische Merkmale der „eisenlastigen Anämien“.“ Der Prototyp ist die β-Thalassämie, eine genetisch rezessive Krankheit, die auf Mutationen des β-Globin-Gens zurückzuführen ist und Anämie und eine übermäßige Produktion der α-Globinkette verursacht. Letztere fällt als Hämichrome im Knochenmark aus, schädigt die heranreifenden erythroiden Vorstufen und führt zu einer ineffektiven Erythropoese. Dies tritt bei nicht transfusionsabhängiger Thalassämie oder Thalassemia intermedia auf, deren Erythropoese durch das Vorhandensein unreifer Zellen gekennzeichnet ist, die Erythroferron freisetzen, um die Hepcidin-Expression in der Leber zu hemmen. Bei transfusionsabhängiger Thalassämie, bei der die endogene ineffektive Erythropoese zumindest teilweise durch Transfusionen unterdrückt wird, sind die Hepcidinspiegel in der Regel höher (Camaschella und Nai, 2016).
Die Hepcidinsuppression wird durch das erhöhte Zytokin Erythroferron (ERFE) vermittelt, ein Mitglied der TNF-α-Familie, das vom ERFE-Gen kodiert wird und von Erythroblasten nach EPO-Stimulation synthetisiert wird (Kautz et al., 2014). ERFE wird in den Blutkreislauf freigesetzt und sequestriert BMPs, insbesondere BMP6 (Arezes et al., 2018), wodurch die Hepcidin-Signalisierung als Reaktion auf Eisen abgeschwächt wird. Darüber hinaus findet am Hepcidin-Locus eine epigenetische Unterdrückung durch die Histon-Deacetylase HDAC3 statt (Pasricha et al., 2017). Wenn Anämie eine Hypoxie verursacht, werden andere Mediatoren wie PDGF-BB (Sonnweber et al., 2014), die von verschiedenen Zelltypen freigesetzt werden, Hepcidin unterdrücken.
Die Hepcidin-Spiegel sind durch einen besonderen Mechanismus bei der Niedrigrisiko-Myelodysplasie mit ringförmigen Sideroblasten, einer klonalen Störung aufgrund von Mutationen des Spleißosomen-Gens SF3B1, erniedrigt. Eisen sammelt sich in Mitochondrien an, was zu einer ineffektiven Erythropoese und einer systemischen Eisenüberladung führt. Ein abnorm gespleißtes, verlängertes ERFE-Protein unterdrückt Hepcidin stärker als der Wildtyp ERFE (Bondu et al., 2019) und verursacht eine transfusionsunabhängige Eisenbelastung.
Zielgerichtete Therapien für Hepcidin-bedingte Anämien
Die Identifizierung der molekularen Mechanismen, die für die zuvor beschriebenen Anämien verantwortlich sind, hat die Forschung zur Entwicklung zielgerichteter Therapien angeregt, um die derzeitige symptomatische Behandlung zu ersetzen (Sebastiani et al., 2016; Crielaard et al., 2017). Die Ansätze unterscheiden sich je nach Art der Anämie und dem Ziel, den Hepcidinspiegel zu senken oder zu erhöhen (Tabelle 2).
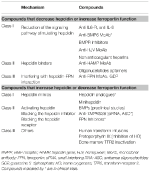
Table 2. Experimentelle Therapien, die auf die Hepcidin-Ferroportin-Achse abzielen.
Experimentelle Therapien zur Senkung des Hepcidin-Spiegels/Steigerung der Ferroportin-Funktion
Abgesehen von Hepcidin-produzierenden Tumoren, die chirurgisch entfernt werden müssen, können Wirkstoffe, die Hepcidin oder seine Wirkungen antagonisieren, bei allen Anämien nützlich sein, die durch hohe Hepcidin-Spiegel gekennzeichnet sind. Sie könnten vor allem bei chronischen Entzündungskrankheiten eingesetzt werden, um Hypoferremie und Anämie zu beseitigen. Mehrere experimentelle Therapien, die auf eine Beeinflussung des Hepcidin-Stoffwechsels und seiner Funktion abzielen, wurden in präklinischen Studien untersucht. Hepcidin-Antagonisten sind Inhibitoren der Hepcidin-Synthese/Regulatoren (Ganz, 2019), Hepcidin-Binder, die seine Funktion blockieren, und Verbindungen, die die Hepcidin-Ferroportin-Interaktion stören (Tabelle 2). Einige Verbindungen befinden sich in klinischen Studien, insbesondere bei chronischen Nierenerkrankungen (Sheetz et al., 2019). Bei IRIDA wurde in präklinischen Studien eine Manipulation des Hepcidin-Stoffwechsels durch den Einsatz von Anti-HJV-MoAb vorgeschlagen (Kovac et al., 2016).
Experimentelle Therapien zur Erhöhung des Hepcidin-Spiegels/zur Verringerung der Ferroportin-Funktion
Eine Erhöhung des Hepcidin-Spiegels kann nicht nur die Eisenüberladung verringern, sondern auch die ineffektive Erythropoese bei eisenbelasteten Anämien teilweise kontrollieren. Die β-Thalassämie ist die am meisten untersuchte dieser Erkrankungen (Casu et al., 2018; Gupta et al., 2018). Vorgeschlagene Medikamente sind Hepcidin-Analoga (einige in klinischen Studien), Hepcidin-Modulatoren, insbesondere TMPRSS6-Inhibitoren, oder Verbindungen, die in die Hepcidin-Ferroportin-Interaktion eingreifen und den Eisenexport verringern (Tabelle 2).
Während Wirkstoffe, die Hepcidin erhöhen, die ineffektive Erythropoese aufgrund des Teufelskreises zwischen ineffektiver Erythropoese und Eisenbelastung reduzieren (Camaschella und Nai, 2016), verbessern Medikamente, die die Reifung der erythroiden Vorläufer begünstigen, wie der Aktivin-Rezeptor-IIB-Ligand Luspatercept, nicht nur die Anämie, sondern auch die Eisenhomöostase, indem sie die Hepcidin-Hemmung reduzieren (Piga et al., 2019).
Einige gezielte Ansätze, die sich derzeit in der klinischen Erprobung befinden, werden hoffentlich zu neuen Behandlungen für eine Vielzahl von Anämien führen.
Schlussfolgerung
Die spektakulären Fortschritte beim Verständnis der Regulierung des Eisenstoffwechsels und des Hepcidins ermöglichten ein besseres Verständnis der Erythropoesekontrolle, da Eisen zusammen mit Erythropoietin ein grundlegender Faktor für die Reifung der Erythrozyten ist. Erkrankungen, die zu Anämie führen, können mit hohen und niedrigen Hepcidinspiegeln einhergehen. In beiden Fällen kann eine gegensätzliche Deregulierung des Hepcidin-Spiegels die Anämie in präklinischen Modellen verbessern/korrigieren und neue Instrumente bieten, die bereits oder bald klinisch für die Behandlung spezifischer Anämien erforscht werden.
Beiträge der Autoren
AP verfasste die Arbeit. CC erstellte die endgültige Fassung. AN und LS trugen zum Schreiben und zur kritischen Durchsicht des Manuskripts bei. Alle Autoren stimmten der Endfassung zu.
Finanzierung
Dieser Beitrag wurde teilweise durch ein EHA Advanced Research Grant 2018 für AP unterstützt. Cariplo Foundation Young Investigator Grant n° 2017-0916 an AN.
Interessenkonflikt
CC ist Berater von Vifor Pharma, Celgene und Novartis.
Die übrigen Autoren erklären, dass die Forschung in Abwesenheit von kommerziellen oder finanziellen Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
An, P., Wu, Q., Wang, H., Guan, Y., Mu, M., Liao, Y., et al. (2012). TMPRSS6, aber nicht TF-, TFR2- oder BMP2-Varianten sind mit einem erhöhten Risiko für Eisenmangelanämie verbunden. Hum. Mol. Genet. 21, 2124-2131. doi: 10.1093/hmg/dds028
PubMed Abstract | CrossRef Full Text | Google Scholar
Andriopoulos, B. Jr., Corradini, E., Xia, Y., Faasse, S. A., Chen, S., Grgurevic, L., et al. (2009). BMP6 ist ein wichtiger endogener Regulator der Hepcidin-Expression und des Eisenstoffwechsels. Nat. Genet. 41, 482-487. doi: 10.1038/ng.335
PubMed Abstract | CrossRef Full Text | Google Scholar
Arezes, J., Foy, N., McHugh, K., Sawant, A., Quinkert, D., Terraube, V., et al. (2018). Erythroferron hemmt die Induktion von Hepcidin durch BMP6. Blood 132, 1473-1477. doi: 10.1182/blood-2018-06-857995
PubMed Abstract | CrossRef Full Text | Google Scholar
Aschemeyer, S., Qiao, B., Stefanova, D., Valore, E. V., Sek, A. C., Ruwe, T. A., et al. (2018). Struktur-Funktions-Analyse von Ferroportin definiert die Bindungsstelle und einen alternativen Wirkmechanismus von Hepcidin. Blood. 131, 899-910.doi: 10.1182/blood-2017-05-786590
CrossRef Full Text | Google Scholar
Babitt, J. L., Huang, F. W., Wrighting, D. M., Xia, Y., Sidis, Y., Samad, T. A., et al. (2006). Knochenmorphogenetisches Protein-Signalisierung durch Hemojuvelin reguliert die Hepcidin-Expression. Nat. Genet. 38, 531-539. doi: 10.1038/ng1777
PubMed Abstract | CrossRef Full Text | Google Scholar
Beguin, Y. (2003). Löslicher Transferrinrezeptor für die Bewertung der Erythropoese und des Eisenstatus. Clin. Chim. Acta 329, 9-22. doi: 10.1016/S0009-8981(03)00005-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Bondu, S., Alary, A. S., Lefevre, C., Houy, A., Jung, G., Lefebvre, T., et al. (2019). Eine Erythroferron-Variante stört die Eisenhomöostase beim SF3B1-mutierten myelodysplastischen Syndrom. Sci. Transl. Med. 11:pii:eaav5467. doi: 10.1126/scitranslmed.aav5467
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C. (2013). Wie ich Patienten mit atypischer mikrozytärer Anämie behandle. Br. J. Haematol. 160, 12-24. doi: 10.1111/bjh.12081
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C. (2019). Iron deficiency. Blood 133, 30-39. doi: 10.1182/blood-2018-05-815944
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C., and Nai, A. (2016). Ineffektive Erythropoese und Regulation des Eisenstatus bei eisenbelasteten Anämien. Br. J. Haematol. 172, 512-523. doi: 10.1111/bjh.13820
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C., and Pagani, A. (2018). Advances in understanding iron metabolism and its crosstalk with erythropoiesis. Br. J. Haematol. 182, 481-494. doi: 10.1111/bjh.15403
PubMed Abstract | CrossRef Full Text | Google Scholar
Canali, S., Wang, C. Y., Zumbrennen-Bullough, K. B., Bayer, A., and Babitt, J. L. (2017). Bone morphogenetic protein 2 controls iron homeostasis in mice independent of Bmp6. Am. J. Hematol. 92, 1204-1213. doi: 10.1002/ajh.24888
PubMed Abstract | CrossRef Full Text | Google Scholar
Casu, C., Nemeth, E., and Rivella, S. (2018). Hepcidin-Agonisten als therapeutische Werkzeuge. Blood 131, 1790-1794. doi: 10.1182/blood-2017-11-737411
PubMed Abstract | CrossRef Full Text | Google Scholar
Colucci, S., Pagani, A., Pettinato, M., Artuso, I., Nai, A., Camaschella, C., et al. (2017). Das Immunophilin FKBP12 hemmt die Hepcidin-Expression durch Bindung des BMP-Typ-I-Rezeptors ALK2 in Hepatozyten. Blood 130, 2111-2120. doi: 10.1182/blood-2017-04-780692
PubMed Abstract | CrossRef Full Text | Google Scholar
Crielaard, B. J., Lammers, T., and Rivella, S. (2017). Targeting iron metabolism in drug discovery and delivery. Nat. Rev. Drug Discov. 16, 400-423. doi: 10.1038/nrd.2016.248
PubMed Abstract | CrossRef Full Text | Google Scholar
De Falco, L., Sanchez, M., Silvestri, L., Kannengiesser, C., Muckenthaler, M. U., Iolascon, A., et al. (2013). Eisenrefraktäre Eisenmangelanämie. Haematologica 98, 845-853. doi: 10.3324/haematol.2012.075515
PubMed Abstract | CrossRef Full Text | Google Scholar
De Falco, L., Silvestri, L., Kannengiesser, C., Moran, E., Oudin, C., Rausa, M., et al. (2014). Funktionelle und klinische Auswirkungen neuer TMPRSS6-Varianten bei Patienten mit eisenrefraktärer Eisenmangelanämie und Genotyp-Phänotyp-Studien. Hum. Mutat. 35, 1321-1329. doi: 10.1002/humu.22632
PubMed Abstract | CrossRef Full Text | Google Scholar
Donker, A. E., Schaap, C. C., Novotny, V. M., Smeets, R., Peters, T. M., van den Heuvel, B. L., et al. (2016). Eisenrefraktäre Eisenmangelanämie: eine heterogene Erkrankung, die nicht immer eisenrefraktär ist. Am. J. Hematol. 91, E482-E490. doi: 10.1002/ajh.24561
PubMed Abstract | CrossRef Full Text | Google Scholar
Du, X., She, E., Gelbart, T., Truksa, J., Lee, P., Xia, Y., et al. (2008). Die Serinprotease TMPRSS6 ist erforderlich, um Eisenmangel zu erkennen. Science 320, 1088-1092. doi: 10.1126/science.1157121
PubMed Abstract | CrossRef Full Text | Google Scholar
Finberg, K. E., Heeney, M. M., Campagna, D. R., Aydinok, Y., Pearson, H. A., Hartman, K. R., et al. (2008). Mutationen in TMPRSS6 verursachen eisenrefraktäre Eisenmangelanämie (IRIDA). Nat. Genet. 40, 569-571. doi: 10.1038/ng.130
PubMed Abstract | CrossRef Full Text | Google Scholar
Ganz, T. (2019). Anemia of Inflammation. N. Engl. J. Med. 381, 1148-1157. doi: 10.1056/NEJMra1804281
PubMed Abstract | CrossRef Full Text | Google Scholar
Gupta, R., Musallam, K. M., Taher, A. T., and Rivella, S. (2018). Ineffektive Erythropoese: Anämie und Eisenüberladung. Hematol. Oncol. Clin. North Am. 32, 213-221. doi: 10.1016/j.hoc.2017.11.009
PubMed Abstract | CrossRef Full Text | Google Scholar
Heeney, M. M., and Finberg, K. E. (2014). Eisenrefraktäre Eisenmangelanämie (IRIDA). Hematol. Oncol. Clin. North Am. 28, 637-652. doi: 10.1016/j.hoc.2014.04.009
PubMed Abstract | CrossRef Full Text | Google Scholar
Heeney, M. M., Guo, D., De Falco, L., Campagna, D. R., Olbina, G., Kao, P. P., et al. (2018). Normalisierendes Hepcidin sagt TMPRSS6-Mutationsstatus bei Patienten mit chronischem Eisenmangel voraus. Blood 132, 448-452. doi: 10.1182/blood-2017-03-773028
PubMed Abstract | CrossRef Full Text | Google Scholar
Kautz, L., Jung, G., Valore, E. V., Rivella, S., Nemeth, E., and Ganz, T. (2014). Identifizierung von Erythroferron als erythroider Regulator des Eisenstoffwechsels. Nat. Genet. 46, 678-684. doi: 10.1038/ng.2996
PubMed Abstract | CrossRef Full Text | Google Scholar
Koch, P. S., Olsavszky, V., Ulbrich, F., Sticht, C., Demory, A., Leibing, T., et al. (2017). Angiocrine Bmp2 signaling in murine liver controls normal iron homeostasis. Blood 129, 415-419. doi: 10.1182/blood-2016-07-729822
PubMed Abstract | CrossRef Full Text | Google Scholar
Kovac, S., Boser, P., Cui, Y., Ferring-Appel, D., Casarrubea, D., Huang, L., et al. (2016). Anti-Hemojuvelin-Antikörper korrigiert Anämie, die durch unangemessen hohe Hepcidin-Spiegel verursacht wird. Haematologica 101, e173-e176. doi: 10.3324/haematol.2015.140772
PubMed Abstract | CrossRef Full Text | Google Scholar
Lakhal, S., Schodel, J., Townsend, A. R., Pugh, C. W., Ratcliffe, P. J., and Mole, D. R. (2011). Regulierung der Typ-II-Transmembran-Serinproteinase TMPRSS6 durch Hypoxie-induzierbare Faktoren: neue Verbindung zwischen Hypoxie-Signalübertragung und Eisenhomöostase. J. Biol. Chem. 286, 4090-4097. doi: 10.1074/jbc.M110.173096
PubMed Abstract | CrossRef Full Text | Google Scholar
Meynard, D., Kautz, L., Darnaud, V., Canonne-Hergaux, F., Coppin, H., and Roth, M. P. (2009). Das Fehlen des morphogenetischen Knochenproteins BMP6 führt zu einer massiven Eisenüberladung. Nat. Genet. 41, 478-481. doi: 10.1038/ng.320
PubMed Abstract | CrossRef Full Text | Google Scholar
Muckenthaler, M. U., Rivella, S., Hentze, M. W., and Galy, B. (2017). Ein roter Teppich für den Eisenstoffwechsel. Cell 168, 344-361. doi: 10.1016/j.cell.2016.12.034
PubMed Abstract | CrossRef Full Text | Google Scholar
Nemeth, E., Tuttle, M. S., Powelson, J., Vaughn, M. B., Donovan, A., Ward, D. M., et al. (2004). Hepcidin reguliert den zellulären Eisen-Efflux durch Bindung an Ferroportin und Induktion seiner Internalisierung. Science 306, 2090-2093. doi: 10.1126/science.1104742
PubMed Abstract | CrossRef Full Text | Google Scholar
Pagani, A., Colucci, S., Bocciardi, R., Bertamino, M., Dufour, C., Ravazzolo, R., et al. (2017). Eine neue Form von IRIDA durch kombinierte heterozygote Mutationen von TMPRSS6 und ACVR1A, die den BMP-Rezeptor ALK2 kodieren. Blood 129, 3392-3395. doi: 10.1182/blood-2017-03-773481
PubMed Abstract | CrossRef Full Text | Google Scholar
Pagani, A., Vieillevoye, M., Nai, A., Rausa, M., Ladli, M., Lacombe, C., et al. (2015). Regulation des Zelloberflächen-Transferrin-Rezeptor-2 durch eisenabhängige Spaltung und Freisetzung einer löslichen Form. Haematologica 100, 458-465. doi: 10.3324/haematol.2014.118521
PubMed Abstract | CrossRef Full Text | Google Scholar
Pasricha, S. R., Lim, P. J., Duarte, T. L., Casu, C., Oosterhuis, D., Mleczko-Sanecka, K., et al. (2017). Hepcidin wird durch Promotor-assoziierte Histon-Acetylierung und HDAC3 reguliert. Nat. Commun. 8:403. doi: 10.1038/s41467-017-00500-z
PubMed Abstract | CrossRef Full Text | Google Scholar
Piga, A., Perrotta, S., Gamberini, M. R., Voskaridou, E., Melpignano, A., Filosa, A., et al. (2019). Luspatercept verbessert die Hämoglobinwerte und den Bluttransfusionsbedarf in einer Studie an Patienten mit Beta-Thalassämie. Blood 133, 1279-1289. doi: 10.1182/blood-2018-10-879247
PubMed Abstract | CrossRef Full Text | Google Scholar
Rausa, M., Pagani, A., Nai, A., Campanella, A., Gilberti, M. E., Apostoli, P., et al. (2015). Bmp6-Expression in nicht parenchymatösen Leberzellen der Maus: ein Mechanismus zur Kontrolle ihrer hohen Eisenexportaktivität und zum Schutz der Hepatozyten vor Eisenüberladung? PLoS One 10:e0122696. doi: 10.1371/journal.pone.0122696
PubMed Abstract | CrossRef Full Text | Google Scholar
Sebastiani, G., Wilkinson, N., and Pantopoulos, K. (2016). Pharmacological targeting of the hepcidin/ferroportin axis. Front. Pharmacol. 7:160. doi: 10.3389/fphar.2016.00160
PubMed Abstract | CrossRef Full Text | Google Scholar
Sheetz, M., Barrington, P., Callies, S., Berg, P. H., McColm, J., Marbury, T., et al. (2019). Targeting the hepcidin-ferroportin pathway in anaemia of chronic kidney disease. Br. J. Clin. Pharmacol. 85, 935-948. doi: 10.1111/bcp.13877
PubMed Abstract | CrossRef Full Text | Google Scholar
Shore, E. M., Xu, M., Feldman, G. J., Fenstermacher, D. A., Cho, T. J., Choi, I. H., et al. (2006). Eine wiederkehrende Mutation im BMP-Typ-I-Rezeptor ACVR1 verursacht vererbte und sporadische Fibrodysplasia ossificans progressiva. Nat. Genet. 38, 525-527. doi: 10.1038/ng1783
PubMed Abstract | CrossRef Full Text | Google Scholar
Silvestri, L., Pagani, A., Nai, A., De Domenico, I., Kaplan, J., and Camaschella, C. (2008). Die Serinprotease Matriptase-2 (TMPRSS6) hemmt die Hepcidin-Aktivierung durch Spaltung von Membran-Hemojuvelin. Cell Metab. 8, 502-511. doi: 10.1016/j.cmet.2008.09.012
PubMed Abstract | CrossRef Full Text | Google Scholar
Silvestri, L., Rausa, M., Pagani, A., Nai, A., and Camaschella, C. (2013). Wie lässt sich die Kausalität von TMPRSS6-Mutationen beurteilen? Hum. Mutat. 34, 1043-1045. doi: 10.1002/humu.22321
PubMed Abstract | CrossRef Full Text | Google Scholar
Sonnweber, T., Nachbaur, D., Schroll, A., Nairz, M., Seifert, M., Demetz, E., et al. (2014). Hypoxia induced downregulation of hepcidin is mediated by platelet derived growth factor BB. Gut 63, 1951-1959. doi: 10.1136/gutjnl-2013-305317
PubMed Abstract | CrossRef Full Text | Google Scholar
Sorensen, E., Rigas, A. S., Didriksen, M., Burgdorf, K. S., Thorner, L. W., Pedersen, O. B., et al. (2019). Genetische Faktoren, die den Hämoglobinspiegel bei 15.567 Blutspendern beeinflussen: Ergebnisse der dänischen Blutspenderstudie. Transfusion 59, 226-231. doi: 10.1111/trf.15075
CrossRef Full Text | Google Scholar
Theurl, I., Schroll, A., Sonnweber, T., Nairz, M., Theurl, M., Willenbacher, W., et al. (2011). Pharmakologische Hemmung der Hepcidin-Expression kehrt Anämie bei chronischer Entzündung in Ratten um. Blood 118, 4977-4984. doi: 10.1182/blood-2011-03-345066
PubMed Abstract | CrossRef Full Text | Google Scholar
Weinstein, D. A., Roy, C. N., Fleming, M. D., Loda, M. F., Wolfsdorf, J. I., and Andrews, N. C. (2002). Unangemessene Expression von Hepcidin ist mit eisenrefraktärer Anämie verbunden: Auswirkungen auf die Anämie bei chronischen Krankheiten. Blood 100, 3776-3781. doi: 10.1182/blood-2002-04-1260
PubMed Abstract | CrossRef Full Text | Google Scholar
Weiss, G., Ganz, T., and Goodnough, L. T. (2019). Anemia of inflammation. Blood 133, 40-50. doi: 10.1182/blood-2018-06-856500
PubMed Abstract | CrossRef Full Text | Google Scholar
Weiss, G., and Goodnough, L. T. (2005). Anämie bei chronischen Krankheiten. N. Engl. J. Med. 352, 1011-1023. doi: 10.1056/NEJMra041809
PubMed Abstract | CrossRef Full Text | Google Scholar
Leave a Reply