Frontières en Physiologie
Introduction
L’anémie est l’un des troubles les plus courants dans le monde et l’anémie due à une carence en fer est la forme prévalente selon de multiples analyses (revue dans Camaschella, 2019). Ce type d’anémie résulte de la carence totale en fer du corps et de l’incapacité à fournir la grande quantité de fer que la moelle osseuse consomme pour produire un nombre adéquat de globules rouges afin de maintenir l’oxygénation des tissus.
La disponibilité du fer est contrôlée par l’hormone peptidique hépatique hepcidine. L’augmentation du fer corporel entraîne la production d’hepcidine, qui est libérée dans la circulation et agit sur son récepteur, la ferroportine, une protéine transmembranaire exportatrice de fer fortement exprimée sur les entérocytes, les macrophages et les hépatocytes. L’hepcidine réduit l’entrée du fer dans le plasma à partir des cellules duodénales absorbantes et des macrophages recycleurs de fer en bloquant l’exportation du fer (Aschemeyer et al., 2018) et en dégradant la ferroportine exportatrice de fer (Nemeth et al., 2004). En régulant le fer plasmatique et l’homéostasie systémique du fer, l’axe hepcidine/ferroportine affecte fortement l’érythropoïèse, d’où le développement possible d’une anémie.
La connexion fer-érythropoïèse
Le processus de production des globules rouges consomme environ 80% du fer circulant pour la synthèse de l’hémoglobine des érythroblastes en maturation. La majeure partie du fer (20-25 mg/jour) est recyclée par les macrophages, tandis qu’une quantité limitée (1-2 mg/jour) provient de l’absorption intestinale. L’hormone rénale érythropoïétine (EPO) contrôle la prolifération des progéniteurs érythroïdes, en particulier des UFC-e et à un degré moindre des UFB-e, et la phase précoce de l’érythropoïèse terminale, tandis que les besoins en fer sont accrus dans les stades tardifs de différenciation des proérythroblastes en réticulocytes, pour la synthèse de l’hème incorporé dans l’hémoglobine (Muckenthaler et al…, 2017).
La régulation de l’hepcidine nécessite une diaphonie entre les cellules sinusoïdales endothéliales du foie (LSEC) qui produisent les protéines morphogénétiques osseuses (BMP) pour activer la voie BMP-SMAD et les hépatocytes qui produisent et libèrent l’hepcidine (Babitt et al., 2006 ; Rausa et al., 2015). BMP6 et BMP2 sont les BMP les plus importantes qui régulent à la hausse l’hepcidine, alors que l’expression de BMP6 est dépendante du fer (Andriopoulos et al., 2009 ; Meynard et al., 2009) BMP2 semble moins sensible au fer (Canali et al., 2017 ; Koch et al., 2017).
Les niveaux d’hepcidine sont bas dans la carence absolue en fer et l’anémie ferriprive. Dans ces conditions, les réserves de fer sont épuisées et la signalisation BMP-SMAD est désactivée à plusieurs niveaux. Tout d’abord, l’expression de BMP6 est supprimée ; ensuite, l’activité de TMPRSS6, une protéase qui clive l’hémojuveline, corécepteur de BMP (Silvestri et al., 2008), est fortement augmentée (Lakhal et al., 2011) ; et enfin, l’histone désacétylase3 (HDAC3) supprime le locus de l’hepcidine (Pasricha et al., 2017). Dans des conditions de carence en fer, la réduction de la production d’hepcidine est un mécanisme d’adaptation qui facilite l’absorption du fer alimentaire et pharmacologique (Camaschella et Pagani, 2018).
Lorsque l’anémie est sévère, l’hypoxie coexistante stimule l’érythropoïèse par une augmentation de la synthèse rénale et de la libération d’EPO. Cela entraîne une suppression de la transcription de l’hepcidine par l’érythroferrone (ERFE), un gène cible de l’EPO produit par les érythroblastes (Kautz et al., 2014), par des molécules (par exemple, PDGF-BB) libérées par d’autres tissus (Sonnweber et al., 2014), et probablement par des composants solubles des récepteurs de la transferrine (TFR), sTFR1 (Beguin, 2003) et sTFR2 (Pagani et al., 2015). L’objectif final est de fournir suffisamment de fer pour les besoins d’une érythropoïèse élargie.
Anémies avec des niveaux d’hepcidine anormaux
Les anémies peuvent être classées sur la base des niveaux d’hepcidine en anémies à forte et faible hepcidine. Il est intuitif que des taux d’hepcidine élevés persistants, en bloquant l’absorption du fer, provoquent une anémie ferriprive en raison de la diminution de l’apport en fer à l’érythropoïèse. À l’inverse, une érythropoïèse inefficace caractérise les anémies dites de charge en fer qui présentent des taux d’hepcidine faibles et une surcharge en fer. Ces deux groupes d’anémies sont le résultat de mécanismes physiopathologiques opposés (figure 1). Dans le premier groupe, l’anémie est due à l’effet inhibiteur exercé par l’hepcidine sur l’absorption et le recyclage du fer qui conduit à une carence systémique en fer ; dans le second groupe, l’anémie est due à la suppression de l’hepcidine par une érythropoïèse anormale étendue (Camaschella et Nai, 2016).
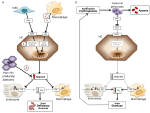
Figure 1. Représentation schématique des mécanismes des anémies à forte (panneau de gauche) et faible hepcidine (panneau de droite). Panneau (A). Pathogénie moléculaire de l’anémie associée à des taux élevés d’hepcidine. LSEC, cellules endothéliales sinusoïdales du foie produisant des protéines morphogénétiques osseuses (BMP) ; BMPRs, récepteurs BMP ; IL6, interleukine 6 ; HC, hépatocytes ; HAMP, gène de l’hepcidine. Fe, fer ; FPN, ferroportine ; 1, IRIDA ; 2, anémie d’inflammation ; 3, adénome producteur d’hepcidine. Panneau (B). Pathogénie moléculaire de la variation de l’hepcidine dans les anémies dues à une érythropoïèse inefficace. ERFE, BMPs séquestrant l’érythroferrone. Les autres mécanismes inhibant l’hepcidine dans ce type d’anémie, comme la diminution de la saturation de la transferrine et l’hypoxie, ne sont pas montrés. Voir le texte pour plus de détails.
Anémie associée à des niveaux élevés d’hepcidine
Ce groupe comprend deux troubles rares héréditaires (anémie ferriprive réfractaire et adénomes producteurs d’hepcidine dans une erreur innée du métabolisme du glucose) et une affection commune acquise : l’anémie d’inflammation (tableau 1).
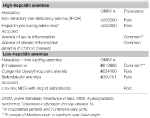
Tableau 1. Anémies classées en fonction des taux d’hepcidine.
Anémie ferriprive réfractaire
L’anémie ferriprive réfractaire (IRIDA) est une maladie récessive rare caractérisée par une anémie microcytaire hypochrome, une faible saturation de la transferrine et des taux d’hepcidine inappropriés normaux/hauts. Elle est causée par des mutations de TMPRSS6 (Finberg et al., 2008), un gène qui code pour la sérine protéase de type II, la matriptase-2 (Du et al., 2008). Les mutations de TMPRSS6 sont réparties le long du gène et peuvent affecter différents domaines, notamment le domaine catalytique (De Falco et al., 2014). Cette protéase transmembranaire, fortement exprimée dans le foie, inhibe la transcription de l’hepcidine en clivant le co-récepteur BMP de surface cellulaire, l’hémojuveline, atténuant ainsi la signalisation BMP et la synthèse de l’hepcidine (Silvestri et al., 2008). La fonction de TMPRSS6 est essentielle dans la carence en fer pour permettre le mécanisme compensatoire d’augmentation de l’absorption du fer.
L’IRDA est présent depuis la naissance et généralement diagnostiqué dans l’enfance. Par rapport à la carence en fer classique, les paramètres du fer sont atypiques et font suspecter la maladie. Le pourcentage de saturation de la transferrine est fortement réduit (moins de 10 %) comme dans les autres formes de carence en fer ; cependant, contrairement à la carence en fer, les niveaux de ferritine sérique sont normaux/augmentés (Camaschella, 2013 ; De Falco et al., 2013). Cela reflète une accumulation accrue de ferritine dans les macrophages, en raison des niveaux élevés d’hepcidine qui induisent la séquestration du fer en magasin.
Aucun des tests proposés pour le diagnostic de l’IRIDA ne couvre 100% des cas. Le test génétique permet d’identifier que les mutations du TMPRSS6, qui dans certains cas (mutations non-sens, frame-shift et épissage), sont clairement causales. Dans d’autres cas, comme pour les mutations faux-sens non signalées auparavant, des études fonctionnelles sont nécessaires pour démontrer la causalité (Silvestri et al., 2013). Or, ces tests sont rarement disponibles. Les taux sériques d’hepcidine sont généralement augmentés/normaux, indépendamment de la carence en fer, et cohérents avec une ferritine élevée/normale. Il est important d’exclure une inflammation en dosant de manière concomitante la protéine C-réactive.
Certains patients présentant un phénotype de carence en fer réfractaire ont été rapportés comme ayant un seul allèle muté TMPRSS6 ; ici, le débat est de savoir s’ils doivent être considérés comme des IRIDA ou non. Un spectre de conditions peut être envisagé, allant de l’IRIDA sévère classique dû à des mutations homozygotes ou hétérozygotes composées de TMPRSS6 à une susceptibilité accrue à la carence en fer conférée par des mutations uniques/changements polymorphes. Une approche proposée pour prédire l’IRIDA classique est la normalisation de l’hepcidine sur d’autres paramètres du fer, comme les ratios saturation de la transferrine (Tsat)/log hepcidine ou Tsat/log Ferritine (Donker et al., 2016). Selon d’autres auteurs, la plupart des patients présentant un phénotype IRIDA sévère présentent des mutations bialéliques de TMPRSS6 et, lorsqu’il n’est pas identifié, le second allèle peut être génétiquement occulte (Heeney et al., 2018). De manière générale, les sujets présentant un seul allèle ont un phénotype plus léger que ceux présentant deux mutations et répondent mieux au traitement par le fer (Donker et al., 2016). Il est intéressant de noter que plusieurs SNP TMPRSS6 se sont avérés conférer une susceptibilité à la carence en fer dans certaines populations (An et al., 2012) et chez les donneurs de sang (Sorensen et al., 2019).
Une hérédité digénique a été rapportée chez une jeune femme de 5 ans qui présentait initialement un génotype IRIDA atypique avec une mutation causale TMPRSS6 (I212T) et une mutation silencieuse (R271Q) (De Falco et al., 2014). Elle a ensuite reçu un diagnostic de Fibrodysplasia ossificans progressiva (FOP), une maladie dominante rare avec une formation osseuse ectopique dans les tissus mous due à la mutation du gène du récepteur BMP de type I ACVR1, codant pour ALK2 (Shore et al., 2006). L’allèle pathologique ALK2R258S est constitutivement actif puisque la mutation affecte le domaine riche en glycine-sérine du gène et rend la voie BMP/SMAD hyperactive étant incapable de lier son inhibiteur spécifique FKBP12 (Pagani et al., 2017).
Ce cas rare est particulièrement illustratif. Premièrement, étant donné que le domaine riche en glycine-sérine d’ALK2 interagit avec FKBP12 et que la mutation déstabilise la liaison, il a révélé un rôle jusqu’alors insoupçonné pour FKBP12 en tant que modulateur d’ALK2 et de l’hepcidine hépatique (Colucci et al., 2017). Deuxièmement, elle a conduit à identifier un lien entre l’activation des récepteurs osseux et hépatiques des BMP de type I. Troisièmement, ce cas renforce la pertinence d’un TMPRSS6 intact dans le contrôle de la signalisation BMP/SMAD hépatique, puisqu’aucun IRIDA n’a été identifié parmi d’autres patients FOP présentant la même mutation ACVR1 et un TMPRSS6 présumé normal (Pagani et al., 2017). Enfin, ce cas est cohérent avec le concept selon lequel l’haploinsuffisance de TMPRSS6 ne peut pas causer d’IRIDA classique.
Le traitement optimal de l’IRIDA n’est pas défini. Le fer oral est inefficace, car il n’est pas absorbé. L’ajout de vitamine C permet une réponse sporadique. Le fer intraveineux induit une réponse partielle, généralement à un rythme plus lent par rapport aux patients présentant une carence en fer acquise. L’EPO est inefficace dans les cas classiques (De Falco et al., 2013 ; Heeney et Finberg, 2014).
Anémie des adénomes producteurs d’hépcidine
C’est une condition extrêmement rare chez les patients adultes affectés par la maladie de stockage du glycogène 1a, un trouble récessif dû à une déficience en glucose-6 phosphatase, qui catalyse une réaction impliquée à la fois dans la glycogénolyse et la gluconéogenèse. Un symptôme dangereux fréquent de la maladie est l’hypoglycémie. Le traitement actuel permet une survie prolongée des enfants atteints jusqu’à l’âge adulte avec l’apparition de plusieurs complications, comme l’anémie et les adénomes hépatiques. L’anémie est microcytaire et hypochrome, carencée en fer et réfractaire au traitement oral par le fer. L’anémie est revenue après la résection chirurgicale de l’adénome. Le tissu de l’adénome s’est révélé positif pour l’ARNm de l’hepcidine, tandis que le tissu normal environnant a montré une suppression de l’hepcidine, comme prévu en raison de la production ectopique incontrôlée d’hepcidine (Weinstein et al., 2002). Les caractéristiques hématologiques des patients ressemblent à celles de l’IRIDA car ils partagent des niveaux élevés d’hepcidine comme mécanisme commun d’anémie.
Anémie de l’inflammation
L’anémie de l’inflammation (AI), précédemment connue sous le nom d’anémie des maladies chroniques, est une anémie normochrome-normocytaire modérée qui se développe dans des conditions d’inflammation systémique et d’activation immunitaire. Elle survient dans plusieurs troubles courants, notamment les infections chroniques, les maladies auto-immunes, les cancers avancés, les maladies rénales chroniques, l’insuffisance cardiaque congestive, les maladies pulmonaires obstructives chroniques, l’anémie des personnes âgées (au moins partiellement) et la maladie du greffon contre l’hôte. L’IA est l’une des anémies les plus courantes dans le monde et l’anémie la plus fréquente chez les patients hospitalisés. L’inflammation aiguë contribue à la gravité de l’anémie dans les unités de soins intensifs. Les mécanismes moléculaires qui sous-tendent l’IA sont multiples et complexes. La surproduction de cytokines telles que l’IL1-β, le TNF-α et l’IL-6 par les macrophages et l’INF-γ par les lymphocytes émousse la production d’EPO, altère la réponse à l’érythropoïèse, augmente les taux d’hepcidine et peut activer l’érythrophagocytose, notamment dans les formes aiguës (Weiss et Goodnough, 2005 ; Ganz, 2019).
L’hepcidine est activée par l’IL-6 via le récepteur de l’IL-6 (IL-6R) et la signalisation JAK2-STAT3. L’activation complète de l’hepcidine nécessite une voie BMP-SMAD active car l’inactivation de la signalisation BMP diminue l’hepcidine dans les modèles animaux d’inflammation (Theurl et al., 2011). La dérégulation de l’homéostasie systémique du fer entraîne une séquestration du fer par les macrophages et une absorption et un recyclage réduits qui conduisent à une faible saturation de la transferrine et à une restriction en fer de l’érythropoïèse et d’autres tissus.
Le traitement traditionnel de l’IA est basé sur la réversibilité/contrôle de la maladie sous-jacente, lorsque cela est possible. Si la maladie n’est pas traitable et que l’anémie est légère, une évaluation minutieuse des risques-bénéfices est nécessaire pour éviter les effets secondaires de tout traitement. Les traitements basés sur la physiopathologie se limitent aux composés de type érythropoïétine et au fer. L’utilisation d’agents stimulant l’érythropoïèse (ASE) supprime l’hepcidine en induisant une expansion de l’érythropoïèse. Cette approche est largement utilisée chez les patients atteints de maladie rénale chronique, de syndromes myélodysplasiques à faible risque et de cancer sous chimiothérapie. Cependant, un contrôle clinique minutieux est nécessaire car les doses élevées ont des effets secondaires cardiovasculaires. L’administration de fer par voie intraveineuse peut soulager la restriction en fer, causée par l’expansion de l’érythropoïèse dépendante des ASE. Le fer oral est généralement inefficace car les taux élevés d’hepcidine s’opposent à son absorption intestinale. Les inhibiteurs de la prolyl hydroxylase (stabilisateurs du facteur inductible de l’hypoxie, HIF) sont expérimentés dans la maladie rénale chronique, dans le but d’augmenter l’EPO endogène. Le traitement chronique par transfusion de globules rouges n’est pas recommandé en raison de l’effet transitoire et des effets indésirables ; il est limité à l’anémie réfractaire sévère (Camaschella, 2019 ; Weiss et al., 2019).
Anémies associées à de faibles niveaux d’hepcidine
L’érythropoïèse inefficace et les niveaux d’hepcidine faibles ou inadéquatement normaux, avec une surcharge en fer conséquente, sont des caractéristiques typiques des » anémies à charge en fer « . » Le prototype est la β-thalassémie, une maladie génétique récessive due à des mutations du gène de la β-globine qui provoquent une anémie et une production excessive de la chaîne d’α-globine. Cette dernière précipite sous forme d’hémichromes dans la moelle osseuse, endommageant les précurseurs érythroïdes en cours de maturation et entraînant une érythropoïèse inefficace. C’est ce qui se produit dans les cas de thalassémie non dépendante des transfusions ou de thalassémie intermédiaire, dont l’érythropoïèse est caractérisée par la prédominance de cellules immatures qui libèrent de l’érythroferrone pour inhiber l’expression de l’hepcidine hépatique. Les taux d’hepcidine sont généralement plus élevés dans les thalassémies dépendantes des transfusions, où l’érythropoïèse inefficace endogène est au moins partiellement supprimée par les transfusions (Camaschella et Nai, 2016).
La suppression de l’hepcidine est médiée par l’augmentation de la cytokine érythroferrone (ERFE), un membre de la famille du TNF-α codé par le gène ERFE, synthétisé par les érythroblastes lors de la stimulation par l’EPO (Kautz et al., 2014). ERFE est libéré dans la circulation et séquestre les BMP, notamment BMP6 (Arezes et al., 2018), atténuant la signalisation de l’hepcidine en réponse au fer. En outre, une suppression épigénétique se produit au niveau du locus de l’hepcidine par l’histone désacétylase HDAC3 (Pasricha et al., 2017). Lorsque l’anémie entraîne une hypoxie, d’autres médiateurs, tels que le PDGF-BB (Sonnweber et al, 2014), qui est libéré par différents types de cellules, suppriment l’hepcidine.
Les niveaux d’hepcidine sont diminués par un mécanisme particulier dans la myélodysplasie à faible risque avec sidéroblastes annelés, un trouble clonal dû à des mutations du gène spliceosome SF3B1. Le fer s’accumule dans les mitochondries, entraînant une érythropoïèse inefficace et une surcharge en fer systémique. Une protéine ERFE allongée et épissée de façon anormale est plus puissante que la protéine ERFE de type sauvage pour supprimer l’hepcidine (Bondu et al., 2019) et en provoquant une charge en fer indépendante des transfusions.
Thérapies ciblées pour les anémies liées à l’hépcidine
L’identification des mécanismes moléculaires responsables des anémies précédemment évoquées a stimulé la recherche dans le développement de thérapies ciblées pour remplacer les traitements symptomatiques actuels (Sebastiani et al., 2016 ; Crielaard et al., 2017). Les approches diffèrent selon le type d’anémie et l’objectif de diminuer ou d’augmenter les niveaux d’hepcidine ou leurs effets (tableau 2).
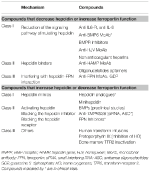
Tableau 2. Thérapies expérimentales ciblant l’axe hepcidine-ferroportine.
Traitements expérimentaux visant à diminuer les niveaux d’hepcidine/accroître la fonction ferroportine
À l’exception des tumeurs produisant de l’hepcidine, qui doivent être retirées chirurgicalement, les composés qui antagonisent l’hepcidine ou ses effets peuvent être utiles dans toutes les anémies caractérisées par des niveaux élevés d’hepcidine. Leur principale application serait dans les maladies inflammatoires chroniques afin d’inverser l’hypoferrémie et l’anémie. Plusieurs thérapies expérimentales visant à manipuler la voie de l’hepcidine et sa fonction ont été étudiées dans des études précliniques. Les antagonistes de l’hepcidine sont des inhibiteurs de la synthèse/régulation de l’hepcidine (Ganz, 2019), des liants de l’hepcidine qui bloquent sa fonction et des composés qui interfèrent avec l’interaction hepcidine-ferroportine (tableau 2). Certains composés sont en cours d’essais cliniques notamment dans les maladies rénales chroniques (Sheetz et al, 2019). Dans l’IRIDA, la manipulation de la voie de l’hepcidine a été proposée dans des études précliniques avec l’utilisation de MoAb anti-HJV (Kovac et al., 2016).
Therapies expérimentales pour augmenter les niveaux d’hepcidine/diminuer la fonction de la ferroportine
L’augmentation des niveaux d’hepcidine peut non seulement réduire la surcharge en fer mais aussi contrôler partiellement l’érythropoïèse inefficace dans les anémies à charge en fer. La β-thalassémie est la plus étudiée parmi ces affections (Casu et al., 2018 ; Gupta et al., 2018). Les médicaments proposés sont des analogues de l’hepcidine (certains en cours d’essais cliniques), des modulateurs de l’hepcidine, en particulier des inhibiteurs du TMPRSS6, ou des composés qui interfèrent avec l’interaction hepcidine-ferroportine diminuant l’exportation du fer (tableau 2).
Alors que les composés qui augmentent l’hepcidine réduisent l’érythropoïèse inefficace en raison du cercle vicieux entre l’érythropoïèse inefficace et la charge en fer (Camaschella et Nai, 2016), les médicaments qui favorisent la maturation des précurseurs érythroïdes, comme le piège ligand du récepteur IIB de l’activine, le luspatercept, non seulement améliorent l’anémie, mais améliorent également l’homéostasie du fer en réduisant l’inhibition de l’hepcidine (Piga et al, 2019).
Certaines approches ciblées qui font maintenant l’objet d’essais cliniques donneront lieu, on l’espère, à de nouveaux traitements pour une variété d’anémies.
Conclusion
Les avancées spectaculaires dans la compréhension de la régulation du métabolisme du fer et de l’hepcidine ont permis de mieux comprendre le contrôle de l’érythropoïèse, car avec l’érythropoïétine, le fer est un facteur fondamental pour la maturation des cellules érythroïdes. Les conditions qui conduisent à l’anémie peuvent être associées à des niveaux élevés et bas d’hepcidine. Dans les deux cas, la dérégulation contrastée de l’hepcidine peut améliorer/corriger l’anémie dans des modèles précliniques, offrant de nouveaux outils qui sont déjà ou seront bientôt explorés cliniquement pour le traitement d’anémies spécifiques.
Contributions des auteurs
AP a rédigé l’article. CC a élaboré la version finale. AN et LS ont contribué à la rédaction et à la révision critique du manuscrit. Tous les auteurs ont approuvé la version finale.
Funding
Cet article a été soutenu en partie par une subvention de recherche avancée de l’EHA en 2018 à AP. Cariplo Foundation Young Investigator Grant n° 2017-0916 à AN.
Conflit d’intérêt
CC est un consultant de Vifor Pharma, Celgene, et Novartis.
Les autres auteurs déclarent que la recherche a été menée en l’absence de toute relation commerciale ou financière qui pourrait être interprétée comme un conflit d’intérêt potentiel.
An, P., Wu, Q., Wang, H., Guan, Y., Mu, M., Liao, Y., et al. (2012). Les variants de TMPRSS6, mais pas ceux de TF, TFR2 ou BMP2 sont associés à un risque accru d’anémie ferriprive. Hum. Mol. Genet. 21, 2124-2131. doi : 10.1093/hmg/dds028
PubMed Abstract | CrossRef Full Text | Google Scholar
Andriopoulos, B. Jr., Corradini, E., Xia, Y., Faasse, S. A., Chen, S., Grgurevic, L., et al. (2009). BMP6 est un régulateur endogène clé de l’expression de l’hepcidine et du métabolisme du fer. Nat. Genet. 41, 482-487. doi : 10.1038/ng.335
PubMed Abstract | CrossRef Full Text | Google Scholar
Arezes, J., Foy, N., McHugh, K., Sawant, A., Quinkert, D., Terraube, V., et al. (2018). L’érythroferrone inhibe l’induction de l’hepcidine par la BMP6. Blood 132, 1473-1477. doi : 10.1182/blood-2018-06-857995
PubMed Abstract | CrossRef Full Text | Google Scholar
Aschemeyer, S., Qiao, B., Stefanova, D., Valore, E. V., Sek, A. C., Ruwe, T. A., et al. (2018). L’analyse structure-fonction de la ferroportine définit le site de liaison et un mécanisme d’action alternatif de l’hepcidine. Blood. 131, 899-910.doi : 10.1182/blood-2017-05-786590
CrossRef Full Text | Google Scholar
Babitt, J. L., Huang, F. W., Wrighting, D. M., Xia, Y., Sidis, Y., Samad, T. A., et al. (2006). Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat. Genet. 38, 531-539. doi : 10.1038/ng1777
PubMed Abstract | CrossRef Full Text | Google Scholar
Beguin, Y. (2003). Récepteur soluble de la transferrine pour l’évaluation de l’érythropoïèse et du statut ferrique. Clin. Chim. Acta 329, 9-22. doi : 10.1016/S0009-8981(03)00005-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Bondu, S., Alary, A. S., Lefevre, C., Houy, A., Jung, G., Lefebvre, T., et al. (2019). Une variante de l’érythroferrone perturbe l’homéostasie du fer dans le syndrome myélodysplasique muté SF3B1. Sci. Transl. Med. 11:pii:eaav5467. doi : 10.1126/scitranslmed.aav5467
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C. (2013). Comment je gère les patients atteints d’anémie microcytaire atypique. Br. J. Haematol. 160, 12-24. doi : 10.1111/bjh.12081
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C. (2019). La carence en fer. Blood 133, 30-39. doi : 10.1182/blood-2018-05-815944
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C., et Nai, A. (2016). Érythropoïèse inefficace et régulation du statut ferrique dans les anémies de charge en fer. Br. J. Haematol. 172, 512-523. doi : 10.1111/bjh.13820
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C., et Pagani, A. (2018). Progrès dans la compréhension du métabolisme du fer et de son crosstalk avec l’érythropoïèse. Br. J. Haematol. 182, 481-494. doi : 10.1111/bjh.15403
PubMed Abstract | CrossRef Full Text | Google Scholar
Canali, S., Wang, C. Y., Zumbrennen-Bullough, K. B., Bayer, A., et Babitt, J. L. (2017). La protéine morphogénétique osseuse 2 contrôle l’homéostasie du fer chez la souris indépendamment de Bmp6. Am. J. Hematol. 92, 1204-1213. doi : 10.1002/ajh.24888
PubMed Abstract | CrossRef Full Text | Google Scholar
Casu, C., Nemeth, E., et Rivella, S. (2018). Les agonistes de l’hépcidine comme outils thérapeutiques. Blood 131, 1790-1794. doi : 10.1182/blood-2017-11-737411
PubMed Abstract | CrossRef Full Text | Google Scholar
Colucci, S., Pagani, A., Pettinato, M., Artuso, I., Nai, A., Camaschella, C., et al. (2017). L’immunophiline FKBP12 inhibe l’expression de l’hepcidine en liant le récepteur BMP de type I ALK2 dans les hépatocytes. Blood 130, 2111-2120. doi : 10.1182/blood-2017-04-780692
PubMed Abstract | CrossRef Full Text | Google Scholar
Crielaard, B. J., Lammers, T., et Rivella, S. (2017). Cibler le métabolisme du fer dans la découverte et l’administration de médicaments. Nat. Rev. Drug Discov. 16, 400-423. doi : 10.1038/nrd.2016.248
PubMed Abstract | CrossRef Full Text | Google Scholar
De Falco, L., Sanchez, M., Silvestri, L., Kannengiesser, C., Muckenthaler, M. U., Iolascon, A., et al. (2013). Anémie ferriprive réfractaire au fer. Haematologica 98, 845-853. doi : 10.3324/haematol.2012.075515
PubMed Abstract | CrossRef Full Text | Google Scholar
De Falco, L., Silvestri, L., Kannengiesser, C., Moran, E., Oudin, C., Rausa, M., et al. (2014). Impact fonctionnel et clinique des nouveaux variants de TMPRSS6 chez les patients atteints d’anémie ferriprive réfractaire et études génotype-phénotype. Hum. Mutat. 35, 1321-1329. doi : 10.1002/humu.22632
PubMed Abstract | CrossRef Full Text | Google Scholar
Donker, A. E., Schaap, C. C., Novotny, V. M., Smeets, R., Peters, T. M., van den Heuvel, B. L., et al. (2016). L’anémie ferriprive réfractaire au fer : une maladie hétérogène qui n’est pas toujours réfractaire au fer. Am. J. Hematol. 91, E482-E490. doi : 10.1002/ajh.24561
PubMed Abstract | CrossRef Full Text | Google Scholar
Du, X., She, E., Gelbart, T., Truksa, J., Lee, P., Xia, Y., et al. (2008). La sérine protéase TMPRSS6 est nécessaire pour détecter la carence en fer. Science 320, 1088-1092. doi : 10.1126/science.1157121
PubMed Abstract | CrossRef Full Text | Google Scholar
Finberg, K. E., Heeney, M. M., Campagna, D. R., Aydinok, Y., Pearson, H. A., Hartman, K. R., et al. (2008). Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA). Nat. Genet. 40, 569-571. doi : 10.1038/ng.130
PubMed Abstract | CrossRef Full Text | Google Scholar
Ganz, T. (2019). L’anémie de l’inflammation. N. Engl. J. Med. 381, 1148-1157. doi : 10.1056/NEJMra1804281
PubMed Abstract | CrossRef Full Text | Google Scholar
Gupta, R., Musallam, K. M., Taher, A. T., et Rivella, S. (2018). Érythropoïèse inefficace : anémie et surcharge en fer. Hematol. Oncol. Clin. North Am. 32, 213-221. doi : 10.1016/j.hoc.2017.11.009
PubMed Abstract | CrossRef Full Text | Google Scholar
Heeney, M. M., et Finberg, K. E. (2014). Anémie ferriprive réfractaire au fer (IRIDA). Hematol. Oncol. Clin. North Am. 28, 637-652. doi : 10.1016/j.hoc.2014.04.009
PubMed Abstract | CrossRef Full Text | Google Scholar
Heeney, M. M., Guo, D., De Falco, L., Campagna, D. R., Olbina, G., Kao, P. P., et al. (2018). L’hepcidine normalisée prédit le statut de la mutation TMPRSS6 chez les patients atteints de carence en fer chronique. Blood 132, 448-452. doi : 10.1182/blood-2017-03-773028
PubMed Abstract | CrossRef Full Text | Google Scholar
Kautz, L., Jung, G., Valore, E. V., Rivella, S., Nemeth, E., et Ganz, T. (2014). Identification de l’érythroferrone comme régulateur érythroïde du métabolisme du fer. Nat. Genet. 46, 678-684. doi : 10.1038/ng.2996
PubMed Abstract | CrossRef Full Text | Google Scholar
Koch, P. S., Olsavszky, V., Ulbrich, F., Sticht, C., Demory, A., Leibing, T., et al. (2017). La signalisation angiocrine Bmp2 dans le foie murin contrôle l’homéostasie normale du fer. Blood 129, 415-419. doi : 10.1182/blood-2016-07-729822
PubMed Abstract | CrossRef Full Text | Google Scholar
Kovac, S., Boser, P., Cui, Y., Ferring-Appel, D., Casarrubea, D., Huang, L., et al. (2016). L’anticorps anti-hémojuveline corrige l’anémie causée par des niveaux d’hepcidine élevés de manière inappropriée. Haematologica 101, e173-e176. doi : 10.3324/haematol.2015.140772
PubMed Abstract | CrossRef Full Text | Google Scholar
Lakhal, S., Schodel, J., Townsend, A. R., Pugh, C. W., Ratcliffe, P. J., et Mole, D. R. (2011). Regulation of type II transmembrane serine proteinase TMPRSS6 by hypoxia-inducible factors : new link between hypoxia signaling and iron homeostasis. J. Biol. Chem. 286, 4090-4097. doi : 10.1074/jbc.M110.173096
PubMed Abstract | CrossRef Full Text | Google Scholar
Meynard, D., Kautz, L., Darnaud, V., Canonne-Hergaux, F., Coppin, H., et Roth, M. P. (2009). L’absence de la protéine morphogénétique osseuse BMP6 induit une surcharge massive en fer. Nat. Genet. 41, 478-481. doi : 10.1038/ng.320
PubMed Abstract | CrossRef Full Text | Google Scholar
Muckenthaler, M. U., Rivella, S., Hentze, M. W., et Galy, B. (2017). Un tapis rouge pour le métabolisme du fer. Cell 168, 344-361. doi : 10.1016/j.cell.2016.12.034
PubMed Abstract | CrossRef Full Text | Google Scholar
Nemeth, E., Tuttle, M. S., Powelson, J., Vaughn, M. B., Donovan, A., Ward, D. M., et al. (2004). Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306, 2090-2093. doi : 10.1126/science.1104742
PubMed Abstract | CrossRef Full Text | Google Scholar
Pagani, A., Colucci, S., Bocciardi, R., Bertamino, M., Dufour, C., Ravazzolo, R., et al. (2017). Une nouvelle forme d’IRIDA due à des mutations hétérozygotes combinées de TMPRSS6 et ACVR1A codant pour le récepteur BMP ALK2. Blood 129, 3392-3395. doi : 10.1182/blood-2017-03-773481
PubMed Abstract | CrossRef Full Text | Google Scholar
Pagani, A., Vieillevoye, M., Nai, A., Rausa, M., Ladli, M., Lacombe, C., et al. (2015). Régulation du récepteur-2 de la transferrine de surface cellulaire par le clivage et la libération d’une forme soluble dépendants du fer. Haematologica 100, 458-465. doi : 10.3324/haematol.2014.118521
PubMed Abstract | CrossRef Full Text | Google Scholar
Pasricha, S. R., Lim, P. J., Duarte, T. L., Casu, C., Oosterhuis, D., Mleczko-Sanecka, K., et al. (2017). L’hépcidine est régulée par l’acétylation des histones associée au promoteur et par HDAC3. Nat. Commun. 8:403. doi : 10.1038/s41467-017-00500-z
PubMed Abstract | CrossRef Full Text | Google Scholar
Piga, A., Perrotta, S., Gamberini, M. R., Voskaridou, E., Melpignano, A., Filosa, A., et al. (2019). Luspatercept améliore les taux d’hémoglobine et les besoins en transfusion sanguine dans une étude de patients atteints de bêta-thalassémie. Blood 133, 1279-1289. doi : 10.1182/blood-2018-10-879247
PubMed Abstract | CrossRef Full Text | Google Scholar
Rausa, M., Pagani, A., Nai, A., Campanella, A., Gilberti, M. E., Apostoli, P., et al. (2015). L’expression de Bmp6 dans les cellules non parenchymateuses du foie murin : un mécanisme pour contrôler leur forte activité d’exportateur de fer et protéger les hépatocytes de la surcharge en fer ? PLoS One 10:e0122696. doi : 10.1371/journal.pone.0122696
PubMed Abstract | CrossRef Full Text | Google Scholar
Sebastiani, G., Wilkinson, N., et Pantopoulos, K. (2016). Ciblage pharmacologique de l’axe hepcidine/ferroportine. Front. Pharmacol. 7:160. doi : 10.3389/fphar.2016.00160
PubMed Abstract | CrossRef Full Text | Google Scholar
Sheetz, M., Barrington, P., Callies, S., Berg, P. H., McColm, J., Marbury, T., et al. (2019). Cibler la voie de l’hepcidine-ferroportine dans l’anémie de la maladie rénale chronique. Br. J. Clin. Pharmacol. 85, 935-948. doi : 10.1111/bcp.13877
PubMed Abstract | CrossRef Full Text | Google Scholar
Shore, E. M., Xu, M., Feldman, G. J., Fenstermacher, D. A., Cho, T. J., Choi, I. H., et al. (2006). A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 38, 525-527. doi : 10.1038/ng1783
PubMed Abstract | CrossRef Full Text | Google Scholar
Silvestri, L., Pagani, A., Nai, A., De Domenico, I., Kaplan, J., et Camaschella, C. (2008). La sérine protéase matriptase-2 (TMPRSS6) inhibe l’activation de l’hepcidine en clivant l’hémojuveline membranaire. Cell Metab. 8, 502-511. doi : 10.1016/j.cmet.2008.09.012
PubMed Abstract | CrossRef Full Text | Google Scholar
Silvestri, L., Rausa, M., Pagani, A., Nai, A., et Camaschella, C. (2013). Comment évaluer la causalité des mutations TMPRSS6 ? Hum. Mutat. 34, 1043-1045. doi : 10.1002/humu.22321
PubMed Abstract | CrossRef Full Text | Google Scholar
Sonnweber, T., Nachbaur, D., Schroll, A., Nairz, M., Seifert, M., Demetz, E., et al. (2014). La dérégulation de l’hepcidine induite par l’hypoxie est médiée par le facteur de croissance dérivé des plaquettes BB. Gut 63, 1951-1959. doi : 10.1136/gutjnl-2013-305317
PubMed Abstract | CrossRef Full Text | Google Scholar
Sorensen, E., Rigas, A. S., Didriksen, M., Burgdorf, K. S., Thorner, L. W., Pedersen, O. B., et al. (2019). Facteurs génétiques influençant les taux d’hémoglobine chez 15 567 donneurs de sang : résultats de l’étude danoise sur les donneurs de sang. Transfusion 59, 226-231. doi : 10.1111/trf.15075
CrossRef Full Text | Google Scholar
Theurl, I., Schroll, A., Sonnweber, T., Nairz, M., Theurl, M., Willenbacher, W., et al. (2011). L’inhibition pharmacologique de l’expression de l’hepcidine renverse l’anémie de l’inflammation chronique chez les rats. Blood 118, 4977-4984. doi : 10.1182/blood-2011-03-345066
PubMed Abstract | CrossRef Full Text | Google Scholar
Weinstein, D. A., Roy, C. N., Fleming, M. D., Loda, M. F., Wolfsdorf, J. I., and Andrews, N. C. (2002). Inappropriate expression of hepcidin is associated with iron refractory anemia : implications for the anemia of chronic disease. Blood 100, 3776-3781. doi : 10.1182/blood-2002-04-1260
PubMed Abstract | CrossRef Full Text | Google Scholar
Weiss, G., Ganz, T., et Goodnough, L. T. (2019). L’anémie de l’inflammation. Blood 133, 40-50. doi : 10.1182/blood-2018-06-856500
PubMed Abstract | CrossRef Full Text | Google Scholar
Weiss, G., et Goodnough, L. T. (2005). L’anémie des maladies chroniques. N. Engl. J. Med. 352, 1011-1023. doi : 10.1056/NEJMra041809
PubMed Abstract | CrossRef Full Text | Google Scholar
Leave a Reply