Frontiers in Physiology
Introduzione
L’anemia è uno dei disturbi più comuni a livello mondiale e l’anemia dovuta a carenza di ferro è la forma prevalente secondo molteplici analisi (review in Camaschella, 2019). Questo tipo di anemia deriva dalla carenza di ferro totale del corpo e dall’incapacità di fornire la grande quantità di ferro che il midollo osseo consuma per produrre un numero adeguato di globuli rossi al fine di mantenere l’ossigenazione dei tessuti.
La disponibilità di ferro è controllata dall’ormone peptidico epatico hepcidin. L’aumento del ferro nel corpo causa la produzione di epcidina, che viene rilasciata nella circolazione e agisce sul suo recettore ferroportina, una proteina transmembrana esportatrice di ferro altamente espressa su enterociti, macrofagi ed epatociti. L’epcidina riduce l’ingresso del ferro nel plasma dalle cellule duodenali assorbenti e dai macrofagi che riciclano il ferro bloccando l’esportazione del ferro (Aschemeyer et al., 2018) e degradando la ferroportina esportatrice di ferro (Nemeth et al., 2004). Regolando il ferro plasmatico e l’omeostasi sistemica del ferro, l’asse epcidina/ferroportina influenza fortemente l’eritropoiesi, quindi il possibile sviluppo di anemia.
La connessione ferro-eritropoiesi
Il processo di produzione dei globuli rossi consuma circa l’80% del ferro circolante per la sintesi di emoglobina degli eritroblasti in maturazione. La maggior parte del ferro (20-25 mg/giorno) è riciclato dai macrofagi, mentre una quantità limitata (1-2 mg al giorno) deriva dall’assorbimento intestinale. L’ormone renale eritropoietina (EPO) controlla la proliferazione dei progenitori eritroidi, soprattutto delle CFU-e e in misura minore delle BFU-e, e la fase iniziale dell’eritropoiesi terminale, mentre il fabbisogno di ferro aumenta nelle fasi di differenziazione tardiva da proeritroblasti a reticolociti, per la sintesi di eme incorporato nell’emoglobina (Muckenthaler et al, 2017).
La regolazione dell’epcidina richiede un crosstalk tra le cellule sinusoidali endoteliali epatiche (LSEC) che producono le proteine morfogenetiche ossee (BMP) per attivare la via BMP-SMAD e gli epatociti che producono e rilasciano l’epcidina (Babitt et al., 2006; Rausa et al., 2015). BMP6 e BMP2 sono le più importanti BMP che upregolano l’epcidina, mentre l’espressione di BMP6 è ferro dipendente (Andriopoulos et al., 2009; Meynard et al., 2009) BMP2 appare meno ferro dipendente (Canali et al., 2017; Koch et al., 2017).
I livelli di epcidina sono bassi nella carenza assoluta di ferro e nell’anemia da carenza di ferro. In queste condizioni, le riserve di ferro sono esaurite e la segnalazione BMP-SMAD è spenta a più livelli. In primo luogo, l’espressione di BMP6 è soppressa; successivamente, l’attività di TMPRSS6, una proteasi che scinde il co-recettore BMP emojuvelina (Silvestri et al., 2008), è fortemente aumentata (Lakhal et al., 2011); e in terzo luogo, l’istone deacetilasi3 (HDAC3) sopprime il locus di epcidina (Pasricha et al., 2017). In condizioni di carenza di ferro, la riduzione della produzione di epcidina è un meccanismo di adattamento che facilita l’assorbimento dietetico e farmacologico del ferro (Camaschella e Pagani, 2018).
Quando l’anemia è grave, la coesistente ipossia stimola l’eritropoiesi attraverso un aumento della sintesi renale e del rilascio di EPO. Questo porta alla soppressione della trascrizione dell’epcidina da parte dell’eritroferrone (ERFE), un gene target dell’EPO prodotto dagli eritroblasti (Kautz et al., 2014), da molecole (ad esempio, PDGF-BB) rilasciate da altri tessuti (Sonnweber et al., 2014), e probabilmente da componenti solubili dei recettori della transferrina (TFR), sTFR1 (Beguin, 2003), e sTFR2 (Pagani et al., 2015). Lo scopo finale è quello di fornire abbastanza ferro per le esigenze di un’eritropoiesi espansa.
Anemie con livelli anormali di epcidina
Le anemie possono essere classificate sulla base dei livelli di epcidina come anemie con epcidina alta e bassa. È intuitivo che livelli di epcidina persistentemente alti, bloccando l’assorbimento del ferro, causano anemia da carenza di ferro a causa della diminuzione dell’apporto di ferro all’eritropoiesi. Al contrario, l’eritropoiesi inefficace caratterizza le cosiddette anemie da carico di ferro che hanno bassi livelli di epcidina e sovraccarico di ferro. Questi due gruppi di anemie sono il risultato di meccanismi fisiopatologici opposti (Figura 1). Nel primo gruppo, l’anemia è dovuta all’effetto inibitorio esercitato dall’epcidina sull’assorbimento e sul riciclo del ferro che porta alla carenza sistemica di ferro; nel secondo gruppo, l’anemia è dovuta alla soppressione dell’epcidina da parte di un’eritropoiesi anomala espansa (Camaschella e Nai, 2016).
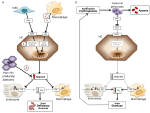
Figura 1. Rappresentazione schematica dei meccanismi di anemie con alta (pannello sinistro) e bassa epcidina (pannello destro). Pannello (A). Patogenesi molecolare dell’anemia associata ad alti livelli di epcidina. LSEC, cellule endoteliali sinusoidali del fegato che producono proteine morfogenetiche ossee (BMPs); BMPRs, recettori BMP; IL6, interleuchina 6; HC, epatociti; HAMP, gene dell’epcidina. Fe, ferro; FPN, ferroportina; 1, IRIDA; 2, anemia di infiammazione; 3, adenoma produttore di epcidina. Pannello (B). Patogenesi molecolare della variazione di epcidina nelle anemie dovute a eritropoiesi inefficace. ERFE, BMP di sequestro dell’eritroferrone. Altri meccanismi che inibiscono l’epcidina in questo tipo di anemia, come la diminuzione della saturazione della transferrina e l’ipossia, non sono mostrati. Vedere il testo per i dettagli.
Anemia associata ad alti livelli di epcidina
Questo gruppo comprende due disturbi rari ereditati (anemia da carenza di ferro refrattaria e adenomi produttori di epcidina in un errore congenito del metabolismo del glucosio) e una condizione comune acquisita: anemia da infiammazione (Tabella 1).
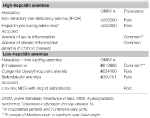
Tabella 1. Anemie classificate secondo i livelli di epcidina.
Anemia da carenza di ferro refrattaria al ferro
L’anemia da carenza di ferro refrattaria al ferro (IRIDA) è un raro disordine recessivo caratterizzato da anemia microcitica ipocromica, bassa saturazione di transferrina e livelli di epcidina inappropriatamente normali/alti. È causato da mutazioni di TMPRSS6 (Finberg et al., 2008), un gene che codifica la serina proteasi di tipo II, matriptasi-2 (Du et al., 2008). Le mutazioni di TMPRSS6 sono diffuse lungo il gene e possono interessare diversi domini, soprattutto il dominio catalitico (De Falco et al., 2014). Questa proteasi transmembrana, altamente espressa nel fegato, inibisce la trascrizione dell’epcidina scindendo il co-recettore BMP di superficie cellulare emojuvelina, attenuando così la segnalazione BMP e la sintesi di epcidina (Silvestri et al., 2008). La funzione di TMPRSS6 è essenziale nella carenza di ferro per consentire il meccanismo compensatorio di aumento dell’assorbimento del ferro.
IRIDA è presente dalla nascita e solitamente diagnosticata nell’infanzia. Rispetto alla carenza di ferro classica, i parametri del ferro sono atipici e sollevano il sospetto della malattia. La percentuale di saturazione della transferrina è fortemente ridotta (meno del 10%) come in altre forme di carenza di ferro; tuttavia, in contrasto con la carenza di ferro, i livelli di ferritina sierica sono normali/aumentati (Camaschella, 2013; De Falco et al., 2013). Questo riflette un maggiore accumulo di ferritina nei macrofagi, a causa degli alti livelli di epcidina che inducono il sequestro del ferro di riserva.
Nessuno dei test proposti per la diagnosi di IRIDA copre il 100% dei casi. Il test genetico identifica che le mutazioni TMPRSS6, che in alcuni casi (mutazioni non-senso, frame-shift e splicing), sono chiaramente causali. In altri casi, come per le mutazioni missense non segnalate in precedenza, sono necessari studi funzionali per dimostrare la causalità (Silvestri et al., 2013). Tuttavia, questi test sono scarsamente disponibili. I livelli sierici di epcidina sono solitamente aumentati/normali, indipendentemente dalla carenza di ferro, e coerenti con la ferritina alta/normale. È importante escludere l’infiammazione dosando concomitantemente la proteina C-reattiva.
Alcuni pazienti con un fenotipo di carenza di ferro refrattaria sono stati segnalati per avere un singolo allele mutato TMPRSS6; qui, il dibattito è se devono essere considerati IRIDA o no. Si può prevedere uno spettro di condizioni che vanno dalla classica IRIDA grave dovuta a mutazioni omozigoti o eterozigoti composti TMPRSS6 ad una maggiore suscettibilità alla carenza di ferro conferita da singole mutazioni / cambiamenti polimorfici. Un approccio proposto per prevedere l’IRIDA classica è la normalizzazione dell’epcidina su altri parametri del ferro, come rapporti saturazione della transferrina (Tsat)/log epcidina o Tsat/log ferritina (Donker et al., 2016). Secondo altri autori, la maggior parte dei pazienti con un fenotipo IRIDA grave ha mutazioni bialleliche di TMPRSS6 e, quando non identificato, il secondo allele può essere geneticamente occulto (Heeney et al., 2018). In termini generali, i soggetti con un singolo allele hanno un fenotipo più lieve di quelli con due mutazioni e rispondono meglio al trattamento con il ferro (Donker et al., 2016). È interessante notare che diversi SNPs di TMPRSS6 hanno dimostrato di fornire suscettibilità alla carenza di ferro in alcune popolazioni (An et al., 2012) e nei donatori di sangue (Sorensen et al., 2019).
È stata riportata un’eredità digenica in una femmina di 5 anni originariamente trovata con un genotipo IRIDA atipico con una mutazione causale TMPRSS6 (I212T) e una (R271Q) silente (De Falco et al., 2014). In seguito le è stata diagnosticata la Fibrodisplasia ossificante progressiva (FOP), un raro disordine dominante con formazione di osso ectopico nei tessuti molli dovuto al gene mutato del recettore BMP di tipo I ACVR1, che codifica ALK2 (Shore et al., 2006). L’allele patologico ALK2R258S è costitutivamente attivo poiché la mutazione colpisce il dominio ricco di glicina-serina del gene e rende il percorso BMP/SMAD iperattivo, essendo incapace di legare il suo inibitore specifico FKBP12 (Pagani et al., 2017).
Questo raro caso è particolarmente illustrativo. In primo luogo, poiché il dominio ALK2 ricco di glicina-serina interagisce con FKBP12 e la mutazione destabilizza il legame, ha rivelato un ruolo precedentemente insospettato per FKBP12 come modulatore di ALK2 epatico e di epcidina (Colucci et al., 2017). In secondo luogo, ha portato a identificare un legame tra l’attivazione dei recettori BMP di tipo I dell’osso e del fegato. In terzo luogo, il caso rafforza la rilevanza della TMPRSS6 intatta nel controllo della segnalazione BMP/SMAD epatica, poiché non è stata identificata alcuna IRIDA tra altri pazienti FOP con la stessa mutazione ACVR1 e presumibilmente TMPRSS6 normale (Pagani et al., 2017). Infine, questo caso è coerente con il concetto che l’aploinsufficienza di TMPRSS6 non può causare IRIDA classica.
Il trattamento ottimale di IRIDA non è definito. Il ferro orale è inefficace, poiché non viene assorbito. L’aggiunta di vitamina C permette una risposta sporadica. Il ferro per via endovenosa induce una risposta parziale di solito ad un ritmo più lento rispetto ai pazienti con carenza di ferro acquisita. L’EPO è inefficace nei casi classici (De Falco et al., 2013; Heeney e Finberg, 2014).
Anemia degli adenomi produttori di epcidina
Questa è una condizione estremamente rara in pazienti adulti affetti dalla malattia da accumulo di glicogeno 1a, un disordine recessivo dovuto alla carenza di glucosio-6 fosfatasi, che catalizza una reazione coinvolta sia nella glicogenolisi che nella gluconeogenesi. Un comune sintomo pericoloso della malattia è l’ipoglicemia. Il trattamento attuale porta ad una sopravvivenza prolungata dei bambini affetti fino all’età adulta con l’insorgenza di diverse complicazioni, come l’anemia e gli adenomi epatici. L’anemia è microcitica e ipocromica, carente di ferro e refrattaria al trattamento orale con ferro. L’anemia è ritornata dopo la resezione chirurgica dell’adenoma. Il tessuto dell’adenoma è stato trovato positivo per l’mRNA dell’epcidina, mentre il tessuto normale circostante ha mostrato la soppressione dell’epcidina, come previsto a causa della produzione ectopica incontrollata di epcidina (Weinstein et al., 2002). Le caratteristiche ematologiche dei pazienti assomigliano a quelle dell’IRIDA in quanto condividono alti livelli di epcidina come meccanismo comune di anemia.
Anemia da infiammazione
L’anemia da infiammazione (AI), precedentemente nota come anemia da malattie croniche, è una moderata anemia normocromica-normocitica che si sviluppa in condizioni di infiammazione sistemica e attivazione immunitaria. Si verifica in diversi disturbi comuni, tra cui infezioni croniche, malattie autoimmuni, cancro avanzato, malattia renale cronica, insufficienza cardiaca congestizia, malattia polmonare ostruttiva cronica, anemia degli anziani (almeno in parte), e graft versus host disease. L’IA è una delle anemie più comuni in tutto il mondo e l’anemia più frequente nei pazienti ospedalizzati. L’infiammazione acuta contribuisce alla gravità dell’anemia nelle unità di terapia intensiva. I meccanismi molecolari alla base dell’IA sono molteplici e complessi. La sovrapproduzione di citochine come IL1-β, TNF-α e IL-6 da parte dei macrofagi e INF-γ da parte dei linfociti interrompe la produzione di EPO, compromette la risposta dell’eritropoiesi, aumenta i livelli di epcidina e può attivare l’eritrofagocitosi, soprattutto nelle forme acute (Weiss e Goodnough, 2005; Ganz, 2019).
L’epcidina è attivata da IL-6 attraverso il recettore IL-6 (IL-6R) e la segnalazione JAK2-STAT3. L’attivazione completa dell’epcidina richiede una via BMP-SMAD attiva perché l’inattivazione della segnalazione BMP diminuisce l’epcidina in modelli animali di infiammazione (Theurl et al., 2011). La deregolazione dell’omeostasi sistemica del ferro causa il sequestro del ferro da parte dei macrofagi e un ridotto assorbimento e riciclo che porta a una bassa saturazione della transferrina e alla restrizione del ferro dell’eritropoiesi e di altri tessuti.
Il trattamento tradizionale dell’IA si basa sulla reversibilità/controllo della malattia sottostante, quando possibile. Se la malattia non è trattabile e l’anemia è lieve, è necessaria un’attenta valutazione dei rischi-benefici per evitare gli effetti collaterali di qualsiasi trattamento. I trattamenti basati sulla fisiopatologia sono limitati a composti simili all’eritropoietina e al ferro. L’uso di agenti stimolanti l’eritropoiesi (ESA) sopprime l’epcidina inducendo l’espansione dell’eritropoiesi. Questo approccio è ampiamente utilizzato nei pazienti con malattia renale cronica, sindromi mielodisplastiche a basso rischio e cancro sottoposti a chemioterapia. Tuttavia, è necessario un attento controllo clinico perché dosi elevate hanno effetti collaterali cardiovascolari. La somministrazione di ferro per via endovenosa può alleviare la restrizione di ferro, causata dall’espansione dell’eritropoiesi dipendente dagli ESA. Il ferro orale è di solito inefficace perché gli alti livelli di epcidina contrastano il suo assorbimento intestinale. Gli inibitori della prolilidrossilasi (hypoxia inducible factor, stabilizzatori HIF) sono sperimentali nella malattia renale cronica, allo scopo di aumentare l’EPO endogena. Il trattamento cronico con trasfusioni di globuli rossi non è raccomandato a causa dell’effetto transitorio e delle reazioni avverse; è limitato alle gravi anemie refrattarie (Camaschella, 2019; Weiss et al., 2019).
Anemie associate a bassi livelli di epcidina
L’eritropoiesi inefficace e livelli di epcidina bassi o inappropriatamente normali, con conseguente sovraccarico di ferro, sono caratteristiche tipiche delle “anemie da carico di ferro”.” Il prototipo è la β-talassemia, una malattia genetica recessiva dovuta a mutazioni del gene della β-globina che causa anemia ed eccesso di produzione della catena α-globina. Quest’ultima precipita come emicromi nel midollo osseo, danneggiando i precursori eritroidi in maturazione e portando a un’eritropoiesi inefficace. Questo si verifica nella talassemia non trasfusione-dipendente o talassemia intermedia, la cui eritropoiesi è caratterizzata dalla prevalenza di cellule immature che rilasciano eritroferrone per inibire l’espressione epatica dell’epcidina. I livelli di epcidina sono solitamente maggiori nella talassemia trasfusione-dipendente, dove l’eritropoiesi inefficace endogena è almeno parzialmente soppressa dalle trasfusioni (Camaschella e Nai, 2016).
La soppressione dell’epcidina è mediata dall’aumento della citochina eritroferrone (ERFE), un membro della famiglia TNF-α codificato dal gene ERFE, sintetizzato dagli eritroblasti su stimolazione EPO (Kautz et al., 2014). ERFE viene rilasciato nella circolazione e sequestra le BMP, soprattutto la BMP6 (Arezes et al., 2018), attenuando la segnalazione dell’epcidina in risposta al ferro. Inoltre, si verifica una soppressione epigenetica al locus dell’epcidina da parte dell’istone deacetilasi HDAC3 (Pasricha et al., 2017). Quando l’anemia causa ipossia, altri mediatori come PDGF-BB (Sonnweber et al, 2014), che viene rilasciato da diversi tipi di cellule, sopprimono l’epcidina.
I livelli di epcidina sono diminuiti da un meccanismo particolare nella mielodisplasia a basso rischio con sideroblasti ad anello, un disordine clonale dovuto a mutazioni del gene dello spliceosoma SF3B1. Il ferro si accumula nei mitocondri, portando a un’eritropoiesi inefficace e a un sovraccarico sistemico di ferro. Una proteina ERFE con splicing anomalo e allungato è più potente di ERFE di tipo selvaggio nel sopprimere l’epcidina (Bondu et al., 2019) e causando un carico di ferro indipendente dalle trasfusioni.
Targeted Therapies for Hepcidin-Related Anemias
L’identificazione dei meccanismi molecolari responsabili delle anemie precedentemente discusse ha stimolato la ricerca nello sviluppo di terapie mirate per sostituire gli attuali trattamenti sintomatici (Sebastiani et al., 2016; Crielaard et al., 2017). Gli approcci differiscono a seconda del tipo di anemia e dell’obiettivo di diminuire o aumentare i livelli di epcidina o i loro effetti (Tabella 2).
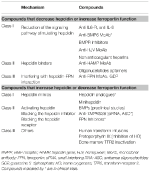
Tabella 2. Terapie sperimentali che mirano all’asse epcidina-ferroportina.
Terapie sperimentali per diminuire i livelli di epcidina/aumentare la funzione della ferroportina
Ad eccezione dei tumori che producono epcidina, che devono essere rimossi chirurgicamente, i composti che antagonizzano l’epcidina o i suoi effetti possono essere utili in tutte le anemie caratterizzate da alti livelli di epcidina. La loro principale applicazione sarebbe nelle malattie infiammatorie croniche per invertire l’ipoferremia e l’anemia. Diverse terapie sperimentali volte a manipolare il percorso dell’epcidina e la sua funzione sono state studiate in studi preclinici. Gli antagonisti dell’epcidina sono inibitori della sintesi/regolazione dell’epcidina (Ganz, 2019), leganti dell’epcidina che bloccano la sua funzione e composti che interferiscono con l’interazione epcidina-ferroportina (Tabella 2). Alcuni composti sono in sperimentazione clinica soprattutto nella malattia renale cronica (Sheetz et al., 2019). Nell’IRIDA, la manipolazione della via dell’epcidina è stata proposta in studi preclinici con l’uso di MoAb anti-HJV (Kovac et al., 2016).
Terapie sperimentali per aumentare i livelli di epcidina/diminuire la funzione della ferroportina
L’aumento dei livelli di epcidina può non solo ridurre il sovraccarico di ferro ma anche controllare parzialmente l’eritropoiesi inefficace nelle anemie da carico di ferro. La β-talassemia è la più studiata tra queste condizioni (Casu et al., 2018; Gupta et al., 2018). I farmaci proposti sono analoghi dell’epcidina (alcuni in studi clinici), modulatori dell’epcidina, specialmente inibitori di TMPRSS6, o composti che interferiscono con l’interazione epcidina-ferroportina diminuendo l’esportazione di ferro (Tabella 2).
Mentre i composti che aumentano l’epcidina riducono l’eritropoiesi inefficace a causa del circolo vizioso tra eritropoiesi inefficace e carico di ferro (Camaschella e Nai, 2016), i farmaci che favoriscono la maturazione dei precursori eritroidi, come la trappola legante del recettore IIB dell’attivina, luspatercept, non solo migliorano l’anemia ma migliorano anche l’omeostasi del ferro riducendo l’inibizione dell’epcidina (Piga et al, 2019).
Alcuni approcci mirati, ora in fase di sperimentazione clinica, si spera possano portare a nuovi trattamenti per una varietà di anemie.
Conclusione
Gli spettacolari progressi nella comprensione della regolazione del metabolismo del ferro e dell’epcidina hanno permesso una migliore comprensione del controllo dell’eritropoiesi, poiché insieme all’eritropoietina il ferro è un fattore fondamentale per la maturazione delle cellule eritroidi. Le condizioni che portano all’anemia possono essere associate a livelli di epcidina alti e bassi. In entrambi i casi, il contrasto della deregolazione dell’epcidina può migliorare/correggere l’anemia in modelli preclinici, offrendo nuovi strumenti che sono già o saranno presto esplorati clinicamente per il trattamento di specifiche anemie.
Contributi degli autori
AP ha redatto l’articolo. CC ha sviluppato la versione finale. AN e LS hanno contribuito alla scrittura e alla revisione critica del manoscritto. Tutti gli autori hanno approvato la versione finale.
Finanziamento
Questo articolo è stato sostenuto in parte da un EHA Advanced Research Grant nel 2018 a AP. Cariplo Foundation Young Investigator Grant n° 2017-0916 a AN.
Conflict of Interest
CC è consulente di Vifor Pharma, Celgene, e Novartis.
I restanti autori dichiarano che la ricerca è stata condotta in assenza di relazioni commerciali o finanziarie che possano essere interpretate come un potenziale conflitto di interessi.
An, P, Wu, Q., Wang, H., Guan, Y., Mu, M., Liao, Y., et al. (2012). TMPRSS6, ma non TF, TFR2 o varianti BMP2 sono associati ad un aumentato rischio di anemia da carenza di ferro. Hum. Mol. Genet. 21, 2124-2131. doi: 10.1093/hmg/dds028
PubMed Abstract | CrossRef Full Text | Google Scholar
Andriopoulos, B. Jr., Corradini, E., Xia, Y., Faasse, S. A., Chen, S., Grgurevic, L., et al. BMP6 è un regolatore endogeno chiave di espressione hepcidin e il metabolismo del ferro. Nat. Genet. 41, 482-487. doi: 10.1038/ng.335
PubMed Abstract | CrossRef Full Text | Google Scholar
Arezes, J., Foy, N., McHugh, K., Sawant, A., Quinkert, D., Terraube, V., et al. (2018). Eritroferrone inibisce l’induzione di epcidina da BMP6. Blood 132, 1473-1477. doi: 10.1182/blood-2018-06-857995
PubMed Abstract | CrossRef Full Text | Google Scholar
Aschemeyer, S., Qiao, B., Stefanova, D., Valore, E. V., Sek, A. C., Ruwe, T. A., et al. (2018). Analisi struttura-funzione di ferroportina definisce il sito di legame e un meccanismo alternativo di azione di epcidina. Blood. 131, 899-910.doi: 10.1182/blood-2017-05-786590
CrossRef Full Text | Google Scholar
Babitt, J. L., Huang, F. W., Wrighting, D. M., Xia, Y., Sidis, Y., Samad, T. A., et al. (2006). Segnalazione di proteina morfogenetica ossea da hemojuvelin regola l’espressione hepcidin. Nat. Genet. 38, 531-539. doi: 10.1038/ng1777
PubMed Abstract | CrossRef Full Text | Google Scholar
Beguin, Y. (2003). Recettore della transferrina solubile per la valutazione dell’eritropoiesi e dello stato del ferro. Clin. Chim. Acta 329, 9-22. doi: 10.1016/S0009-8981(03)00005-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Bondu, S., Alary, A. S., Lefevre, C., Houy, A., Jung, G., Lefebvre, T., et al. (2019). Una variante eritroferrone interrompe l’omeostasi del ferro nella sindrome mielodisplastica SF3B1-mutata. Sci. Transl. Med. 11:pii:eaav5467. doi: 10.1126/scitranslmed.aav5467
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C. (2013). Come gestire i pazienti con anemia microcitica atipica. Br. J. Haematol. 160, 12-24. doi: 10.1111/bjh.12081
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C. (2019). Carenza di ferro. Blood 133, 30-39. doi: 10.1182/blood-2018-05-815944
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C., and Nai, A. (2016). Eritropoiesi inefficace e regolazione dello stato del ferro nelle anemie da carico di ferro. Br. J. Haematol. 172, 512-523. doi: 10.1111/bjh.13820
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C., e Pagani, A. (2018). Progressi nella comprensione del metabolismo del ferro e del suo crosstalk con l’eritropoiesi. Br. J. Haematol. 182, 481-494. doi: 10.1111/bjh.15403
PubMed Abstract | CrossRef Full Text | Google Scholar
Canali, S., Wang, C. Y., Zumbrennen-Bullough, K. B., Bayer, A., e Babitt, J. L. (2017). La proteina morfogenetica ossea 2 controlla l’omeostasi del ferro nei topi indipendentemente da Bmp6. Am. J. Hematol. 92, 1204-1213. doi: 10.1002/ajh.24888
PubMed Abstract | CrossRef Full Text | Google Scholar
Casu, C., Nemeth, E., e Rivella, S. (2018). Agonisti dell’epcidina come strumenti terapeutici. Blood 131, 1790-1794. doi: 10.1182/blood-2017-11-737411
PubMed Abstract | CrossRef Full Text | Google Scholar
Colucci, S., Pagani, A., Pettinato, M., Artuso, I., Nai, A., Camaschella, C., et al. (2017). L’immunofilina FKBP12 inibisce l’espressione dell’epcidina legando il recettore BMP di tipo I ALK2 negli epatociti. Blood 130, 2111-2120. doi: 10.1182/blood-2017-04-780692
PubMed Abstract | CrossRef Full Text | Google Scholar
Crielaard, B. J., Lammers, T., e Rivella, S. (2017). Targeting metabolismo del ferro nella scoperta della droga e la consegna. Nat. Rev. Drug Discov. 16, 400-423. doi: 10.1038/nrd.2016.248
PubMed Abstract | CrossRef Full Text | Google Scholar
De Falco, L., Sanchez, M., Silvestri, L., Kannengiesser, C., Muckenthaler, M. U., Iolascon, A., et al. (2013). Anemia da carenza di ferro refrattaria. Haematologica 98, 845-853. doi: 10.3324/haematol.2012.075515
PubMed Abstract | CrossRef Full Text | Google Scholar
De Falco, L., Silvestri, L., Kannengiesser, C., Moran, E., Oudin, C., Rausa, M., et al. (2014). Impatto funzionale e clinico di nuove varianti TMPRSS6 in pazienti con anemia da carenza di ferro refrattaria e studi genotipo-fenotipo. Hum. Mutat. 35, 1321-1329. doi: 10.1002/humu.22632
PubMed Abstract | CrossRef Full Text | Google Scholar
Donker, A. E., Schaap, C. C., Novotny, V. M., Smeets, R., Peters, T. M., van den Heuvel, B. L., et al. (2016). Anemia da carenza di ferro refrattaria: una malattia eterogenea che non è sempre refrattaria al ferro. Am. J. Hematol. 91, E482-E490. doi: 10.1002/ajh.24561
PubMed Abstract | CrossRef Full Text | Google Scholar
Du, X., She, E., Gelbart, T., Truksa, J., Lee, P., Xia, Y., et al. (2008). La serina proteasi TMPRSS6 è necessaria per sentire la carenza di ferro. Scienza 320, 1088-1092. doi: 10.1126/science.1157121
PubMed Abstract | CrossRef Full Text | Google Scholar
Finberg, K. E., Heeney, M. M., Campagna, D. R., Aydinok, Y., Pearson, H. A., Hartman, K. R., et al. Mutazioni in TMPRSS6 causare ferro-refrattario anemia da carenza di ferro (IRIDA). Nat. Genet. 40, 569-571. doi: 10.1038/ng.130
PubMed Abstract | CrossRef Full Text | Google Scholar
Ganz, T. (2019). Anemia di infiammazione. N. Engl. J. Med. 381, 1148-1157. doi: 10.1056/NEJMra1804281
PubMed Abstract | CrossRef Full Text | Google Scholar
Gupta, R., Musallam, K. M., Taher, A. T., e Rivella, S. (2018). Eritropoiesi inefficace: anemia e sovraccarico di ferro. Hematol. Oncol. Clin. North Am. 32, 213-221. doi: 10.1016/j.hoc.2017.11.009
PubMed Abstract | CrossRef Full Text | Google Scholar
Heeney, M. M., e Finberg, K. E. (2014). Anemia da carenza di ferro refrattaria (IRIDA). Hematol. Oncol. Clin. North Am. 28, 637-652. doi: 10.1016/j.hoc.2014.04.009
PubMed Abstract | CrossRef Full Text | Google Scholar
Heeney, M. M., Guo, D., De Falco, L., Campagna, D. R., Olbina, G., Kao, P. P., et al. (2018). La normalizzazione dell’epcidina predice lo stato di mutazione TMPRSS6 nei pazienti con carenza cronica di ferro. Blood 132, 448-452. doi: 10.1182/blood-2017-03-773028
PubMed Abstract | CrossRefef Text | Google Scholar
Kautz, L., Jung, G., Valore, E. V., Rivella, S., Nemeth, E., e Ganz, T. (2014). Identificazione di eritroferrone come un regolatore eritroide del metabolismo del ferro. Nat. Genet. 46, 678-684. doi: 10.1038/ng.2996
PubMed Abstract | CrossRef Full Text | Google Scholar
Koch, P. S., Olsavszky, V., Ulbrich, F., Sticht, C., Demory, A., Leibing, T., et al. (2017). Segnalazione angiocrina Bmp2 nel fegato murino controlla normale omeostasi del ferro. Blood 129, 415-419. doi: 10.1182/blood-2016-07-729822
PubMed Abstract | CrossRef Full Text | Google Scholar
Kovac, S., Boser, P., Cui, Y., Ferring-Appel, D., Casarrubea, D., Huang, L., et al. (2016). L’anticorpo anti-hemojuvelin corregge l’anemia causata da livelli di epcidina inappropriatamente alti. Haematologica 101, e173-e176. doi: 10.3324/haematol.2015.140772
PubMed Abstract | CrossRef Full Text | Google Scholar
Lakhal, S., Schodel, J., Townsend, A. R., Pugh, C. W., Ratcliffe, P. J., e Mole, D. R. (2011). Regolazione di tipo II transmembrana serina proteinasi TMPRSS6 da fattori ipossia-inducibili: nuovo legame tra segnalazione ipossia e omeostasi del ferro. J. Biol. Chem. 286, 4090-4097. doi: 10.1074/jbc.M110.173096
PubMed Abstract | CrossRef Full Text | Google Scholar
Meynard, D., Kautz, L., Darnaud, V., Canonne-Hergaux, F., Coppin, H., e Roth, M. P. (2009). La mancanza della proteina morfogenetica ossea BMP6 induce un massiccio sovraccarico di ferro. Nat. Genet. 41, 478-481. doi: 10.1038/ng.320
PubMed Abstract | CrossRef Full Text | Google Scholar
Muckenthaler, M. U., Rivella, S., Hentze, M. W., e Galy, B. (2017). Un tappeto rosso per il metabolismo del ferro. Cell 168, 344-361. doi: 10.1016/j.cell.2016.12.034
PubMed Abstract | CrossRef Full Text | Google Scholar
Nemeth, E., Tuttle, M. S., Powelson, J., Vaughn, M. B., Donovan, A., Ward, D. M., et al. (2004). Hepcidin regola efflusso di ferro cellulare legandosi a ferroportin e inducendo la sua internalizzazione. Scienza 306, 2090-2093. doi: 10.1126/science.1104742
PubMed Abstract | CrossRef Full Text | Google Scholar
Pagani, A., Colucci, S., Bocciardi, R., Bertamino, M., Dufour, C., Ravazzolo, R., et al. (2017). Una nuova forma di IRIDA dovuta a mutazioni combinate eterozigoti di TMPRSS6 e ACVR1A che codifica il recettore BMP ALK2. Blood 129, 3392-3395. doi: 10.1182/blood-2017-03-773481
PubMed Abstract | CrossRef Full Text | Google Scholar
Pagani, A, Vieillevoye, M., Nai, A., Rausa, M., Ladli, M., Lacombe, C., et al. (2015). Regolazione della superficie cellulare transferrina recettore-2 da ferro-dipendente scissione e il rilascio di una forma solubile. Haematologica 100, 458-465. doi: 10.3324/haematol.2014.118521
PubMed Abstract | CrossRef Full Text | Google Scholar
Pasricha, S. R., Lim, P. J., Duarte, T. L., Casu, C., Oosterhuis, D., Mleczko-Sanecka, K., et al. (2017). Hepcidin è regolato da acetilazione istone associato al promotore e HDAC3. Nat. Commun. 8:403. doi: 10.1038/s41467-017-00500-z
PubMed Abstract | CrossRef Full Text | Google Scholar
Piga, A., Perrotta, S., Gamberini, M. R., Voskaridou, E., Melpignano, A., Filosa, A., et al. (2019). Luspatercept migliora i livelli di emoglobina e il fabbisogno di trasfusioni di sangue in uno studio su pazienti con beta-talassemia. Blood 133, 1279-1289. doi: 10.1182/blood-2018-10-879247
PubMed Abstract | CrossRef Full Text | Google Scholar
Rausa, M., Pagani, A., Nai, A., Campanella, A., Gilberti, M. E., Apostoli, P., et al. (2015). Espressione di Bmp6 in cellule epatiche non parenchimali murine: un meccanismo per controllare la loro elevata attività di esportatore di ferro e proteggere gli epatociti dal sovraccarico di ferro? PLoS One 10:e0122696. doi: 10.1371/journal.pone.0122696
PubMed Abstract | CrossRef Full Text | Google Scholar
Sebastiani, G., Wilkinson, N., e Pantopoulos, K. (2016). Targeting farmacologico dell’asse epcidina/ferroportina. Front. Pharmacol. 7:160. doi: 10.3389/fphar.2016.00160
PubMed Abstract | CrossRef Full Text | Google Scholar
Sheetz, M., Barrington, P., Callies, S., Berg, P. H., McColm, J., Marbury, T., et al. (2019). Targeting del percorso hepcidin-ferroportin nell’anemia della malattia renale cronica. Br. J. Clin. Pharmacol. 85, 935-948. doi: 10.1111/bcp.13877
PubMed Abstract | CrossRef Full Text | Google Scholar
Shore, E. M., Xu, M., Feldman, G. J., Fenstermacher, D. A., Cho, T. J., Choi, I. H., et al. (2006). Una mutazione ricorrente nel recettore BMP tipo I ACVR1 causa fibrodisplasia ossificante progressiva ereditata e sporadica. Nat. Genet. 38, 525-527. doi: 10.1038/ng1783
PubMed Abstract | CrossRef Full Text | Google Scholar
Silvestri, L., Pagani, A., Nai, A., De Domenico, I., Kaplan, J., e Camaschella, C. (2008). La serina proteasi matriptasi-2 (TMPRSS6) inibisce l’attivazione dell’epcidina scindendo l’emojuvelina di membrana. Cell Metab. 8, 502-511. doi: 10.1016/j.cmet.2008.09.012
PubMed Abstract | CrossRef Full Text | Google Scholar
Silvestri, L., Rausa, M., Pagani, A., Nai, A., e Camaschella, C. (2013). Come valutare la causalità delle mutazioni TMPRSS6? Hum. Mutat. 34, 1043-1045. doi: 10.1002/humu.22321
PubMed Abstract | CrossRef Full Text | Google Scholar
Sonnweber, T., Nachbaur, D., Schroll, A., Nairz, M., Seifert, M., Demetz, E., et al. (2014). Ipossia indotta downregulation di hepcidin è mediata dal fattore di crescita piastrinico BB. Gut 63, 1951-1959. doi: 10.1136/gutjnl-2013-305317
PubMed Abstract | CrossRef Full Text | Google Scholar
Sorensen, E., Rigas, A. S., Didriksen, M., Burgdorf, K. S., Thorner, L. W., Pedersen, O. B., et al. (2019). Fattori genetici che influenzano i livelli di emoglobina in 15.567 donatori di sangue: risultati dello studio danese sui donatori di sangue. Transfusion 59, 226-231. doi: 10.1111/trf.15075
CrossRef Full Text | Google Scholar
Theurl, I., Schroll, A., Sonnweber, T., Nairz, M., Theurl, M., Willenbacher, W., et al. (2011). Inibizione farmacologica di espressione hepcidin inverte l’anemia di infiammazione cronica nei ratti. Blood 118, 4977-4984. doi: 10.1182/blood-2011-03-345066
PubMed Abstract | CrossRef Full Text | Google Scholar
Weinstein, D. A., Roy, C. N., Fleming, M. D., Loda, M. F., Wolfsdorf, J. I., e Andrews, N. C. (2002). Espressione inappropriata di hepcidin è associato con anemia refrattaria al ferro: implicazioni per l’anemia della malattia cronica. Blood 100, 3776-3781. doi: 10.1182/blood-2002-04-1260
PubMed Abstract | CrossRef Full Text | Google Scholar
Weiss, G., Ganz, T., and Goodnough, L. T. (2019). Anemia di infiammazione. Blood 133, 40-50. doi: 10.1182/blood-2018-06-856500
PubMed Abstract | CrossRef Full Text | Google Scholar
Weiss, G., and Goodnough, L. T. (2005). Anemia della malattia cronica. N. Engl. J. Med. 352, 1011-1023. doi: 10.1056/NEJMra041809
PubMed Abstract | CrossRef Full Text | Google Scholar
Leave a Reply