Frontiers in Physiology
Úvod
Anémie je jedním z nejčastějších onemocnění na světě a anémie způsobená nedostatkem železa je podle mnoha analýz její převažující formou (přehled v Camaschella, 2019). Tento typ anémie je důsledkem celkového nedostatku železa v organismu a neschopnosti dodat velké množství železa, které kostní dřeň spotřebovává k produkci dostatečného počtu červených krvinek pro udržení okysličení tkání.
Dostupnost železa je řízena jaterním peptidovým hormonem hepcidinem. Zvýšení množství železa v těle způsobuje produkci hepcidinu, který se uvolňuje do oběhu a působí na svůj receptor ferroportin, transmembránový protein exportující železo vysoce exprimovaný na enterocytech, makrofázích a hepatocytech. Hepcidin snižuje vstup železa do plazmy z absorpčních duodenálních buněk a makrofágů recyklujících železo tím, že blokuje export železa (Aschemeyer et al., 2018) a degraduje ferroportin exportující železo (Nemeth et al., 2004). Regulací plazmatického železa a systémové homeostázy železa osa hepcidin/ferroportin silně ovlivňuje erytropoézu, a tím i možný rozvoj anémie.
Spojení železa a erytropoézy
Proces tvorby červených krvinek spotřebuje přibližně 80 % cirkulujícího železa na syntézu hemoglobinu zrajících erytroblastů. Většina železa (20-25 mg/den) je recyklována makrofágy, zatímco omezené množství (1-2 mg denně) pochází ze střevní absorpce. Ledvinový hormon erytropoetin (EPO) řídí proliferaci erytroidních progenitorů, zejména CFU-e a v nižší míře BFU-e, a časnou fázi terminální erytropoézy, zatímco potřeba železa se zvyšuje v pozdních fázích diferenciace z proerytroblastů na retikulocyty, pro syntézu hemu inkorporovaného do hemoglobinu (Muckenthaler et al.,
Regulace hepcidinu vyžaduje vzájemnou interakci mezi jaterními endoteliálními sinusoidálními buňkami (LSEC), které produkují kostní morfogenetické proteiny (BMP) k aktivaci dráhy BMP-SMAD, a hepatocyty, které produkují a uvolňují hepcidin (Babitt et al., 2006; Rausa et al., 2015). BMP6 a BMP2 jsou nejdůležitější BMP, které regulují hepcidin, zatímco exprese BMP6 je závislá na železe (Andriopoulos et al., 2009; Meynard et al., 2009), BMP2 se zdá být méně závislý na železe (Canali et al., 2017; Koch et al., 2017).
Hladiny hepcidinu jsou nízké při absolutním nedostatku železa a anémii z nedostatku železa. Za těchto stavů jsou zásoby železa vyčerpány a signalizace BMP-SMAD je vypnuta na více úrovních. Zaprvé je potlačena exprese BMP6, dále je silně zvýšena aktivita TMPRSS6, proteázy, která štěpí ko-receptor BMP hemojuvelin (Silvestri et al., 2008) (Lakhal et al., 2011), a zatřetí histondeacetyláza3 (HDAC3) potlačuje lokus hepcidinu (Pasricha et al., 2017). V podmínkách nedostatku železa je snížení produkce hepcidinu adaptačním mechanismem, který usnadňuje vstřebávání železa stravou a farmakologicky (Camaschella a Pagani, 2018).
Při závažné anémii stimuluje koexistující hypoxie erytropoézu prostřednictvím zvýšené syntézy a uvolňování EPO ledvinami. To vede k potlačení transkripce hepcidinu erytroferronem (ERFE), cílovým genem EPO produkovaným erytroblasty (Kautz et al., 2014), molekulami (např. PDGF-BB) uvolňovanými jinými tkáněmi (Sonnweber et al., 2014) a pravděpodobně i rozpustnými složkami transferinových receptorů (TFR), sTFR1 (Beguin, 2003) a sTFR2 (Pagani et al., 2015). Konečným cílem je dodat dostatek železa pro potřeby rozšířené erytropoézy.
Anémie s abnormální hladinou hepcidinu
Anémie lze na základě hladiny hepcidinu klasifikovat jako anémie s vysokou a nízkou hladinou hepcidinu. Je intuitivní, že trvale vysoké hladiny hepcidinu tím, že blokují vstřebávání železa, způsobují anémii z nedostatku železa z důvodu sníženého přísunu železa do erytropoézy. Naopak neefektivní erytropoéza charakterizuje tzv. anémie s nízkou hladinou hepcidinu a přetížením železem. Tyto dvě skupiny anémií jsou výsledkem opačných patofyziologických mechanismů (obrázek 1). V první skupině je anémie způsobena inhibičním účinkem, který má hepcidin na absorpci a recyklaci železa, což vede k systémovému nedostatku železa; ve druhé skupině je anémie způsobena supresí hepcidinu rozšířenou abnormální erytropoézou (Camaschella a Nai, 2016).
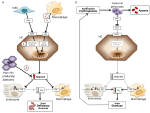
Obrázek 1. Schematické znázornění mechanismů anémií s vysokou (levý panel) a nízkou hladinou hepcidinu (pravý panel). Panel (A). Molekulární patogeneze anémie spojené s vysokou hladinou hepcidinu. LSEC, jaterní sinusoidální endotelové buňky produkující kostní morfogenetické proteiny (BMP); BMPR, receptory BMP; IL6, interleukin 6; HC, hepatocyty; HAMP, gen pro hepcidin. Fe, železo; FPN, feroportin; 1, IRIDA; 2, zánětlivá anémie; 3, adenom produkující hepcidin. Panel (B). Molekulární patogeneze variability hepcidinu u anémií způsobených neefektivní erytropoézou. ERFE, erytroferron sekvestrující BMP. Další mechanismy inhibující hepcidin u tohoto typu anémie, jako snížení saturace transferinem a hypoxie, nejsou zobrazeny. Podrobnosti viz text.
Anémie spojená s vysokou hladinou hepcidinu
Tato skupina zahrnuje dvě dědičné vzácné poruchy (anémie z nedostatku železa refrakterní na železo a adenomy produkující hepcidin při vrozené chybě metabolismu glukózy) a získaný běžný stav: zánětlivou anémii (tabulka 1).
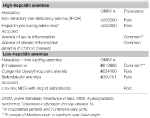
Tabulka 1. Anémie klasifikované podle hladiny hepcidinu.
Anémie z nedostatku železa refrakterní na železo
Anémie z nedostatku železa refrakterní na železo (IRIDA) je vzácná recesivní porucha charakterizovaná hypochromní mikrocytární anémií, nízkou saturací transferinu a nepřiměřeně normální/vysokou hladinou hepcidinu. Je způsobena mutacemi genu TMPRSS6 (Finberg et al., 2008), který kóduje serinovou proteázu typu II, matriptázu-2 (Du et al., 2008). Mutace TMPRSS6 se šíří podél genu a mohou postihovat různé domény, zejména katalytickou doménu (De Falco et al., 2014). Tato transmembránová proteáza, vysoce exprimovaná v játrech, inhibuje transkripci hepcidinu tím, že štěpí ko-receptor BMP na povrchu buněk hemojuvelin, čímž oslabuje signalizaci BMP a syntézu hepcidinu (Silvestri et al., 2008). Funkce TMPRSS6 je nezbytná při nedostatku železa, aby umožnila kompenzační mechanismus zvýšené absorpce železa.
IRIDA je přítomna od narození a obvykle je diagnostikována v dětství. Ve srovnání s klasickým nedostatkem železa jsou parametry železa atypické a vyvolávají podezření na toto onemocnění. Procentuální saturace transferinu je silně snížená (méně než 10 %) jako u jiných forem nedostatku železa; na rozdíl od nedostatku železa jsou však hladiny sérového feritinu normální/zvýšené (Camaschella, 2013; De Falco et al., 2013). To odráží zvýšenou akumulaci feritinu v makrofázích v důsledku vysokých hladin hepcidinu, které vyvolávají sekvestraci zásobního železa.
Žádný z testů navržených pro diagnostiku IRIDA nepokrývá 100 % případů. Genetický test identifikuje mutace TMPRSS6, které jsou v některých případech (non-sense, frame-shift a sestřihové mutace) jednoznačně kauzální. V jiných případech, jako v případě dříve nezaznamenaných missense mutací, je k prokázání kauzality zapotřebí funkčních studií (Silvestri et al., 2013). Tyto testy jsou však jen zřídka dostupné. Sérové hladiny hepcidinu jsou obvykle zvýšené/normální, nezávisle na nedostatku železa, a odpovídají vysokému/normálnímu feritinu. Důležité je vyloučit zánět současným dávkováním C-reaktivního proteinu.
U některých pacientů s fenotypem refrakterního nedostatku železa byla zjištěna jediná mutovaná alela TMPRSS6; zde se diskutuje o tom, zda by měli být považováni za IRIDA, nebo ne. Lze předpokládat spektrum stavů od klasické těžké IRIDA způsobené homozygotními nebo složenými heterozygotními mutacemi TMPRSS6 až po zvýšenou náchylnost k nedostatku železa danou jednotlivými mutacemi/polymorfními změnami. Jedním z přístupů navržených k predikci klasické IRIDA je normalizace hepcidinu na jiné parametry železa, jako poměry saturace transferinu (Tsat)/log hepcidinu nebo Tsat/log feritinu (Donker et al., 2016). Podle jiných autorů má většina pacientů s těžkým fenotypem IRIDA bialelické mutace TMPRSS6, a pokud nejsou identifikovány, může být druhá alela geneticky okultní (Heeney et al., 2018). Obecně lze říci, že osoby s jednou alelou mají mírnější fenotyp než osoby se dvěma mutacemi a lépe reagují na léčbu železem (Donker et al., 2016). Zajímavé je, že u některých populací (An et al., 2012) a u dárců krve (Sorensen et al., 2019) bylo prokázáno, že několik SNP TMPRSS6 poskytuje náchylnost k nedostatku železa.
Byla popsána digenická dědičnost u pětileté dívky, u které byl původně zjištěn atypický genotyp IRIDA s jednou kauzální a jednou tichou mutací TMPRSS6 (I212T) (R271Q) (De Falco et al., 2014). Později jí byla diagnostikována Fibrodysplasia ossificans progressiva (FOP), vzácná dominantní porucha s ektopickou tvorbou kosti v měkkých tkáních v důsledku mutace genu ACVR1 pro receptor BMP typu I, který kóduje ALK2 (Shore et al., 2006). Patologická alela ALK2R258S je konstitutivně aktivní, protože mutace postihuje doménu genu bohatou na glycin a serin a způsobuje, že dráha BMP/SMAD je hyperaktivní, protože není schopna vázat svůj specifický inhibitor FKBP12 (Pagani et al., 2017).
Tento vzácný případ je obzvláště ilustrativní. Za prvé, jelikož doména ALK2 bohatá na glycin a serin interaguje s FKBP12 a mutace destabilizuje vazbu, odhalila dříve netušenou roli FKBP12 jako modulátoru jaterní ALK2 a hepcidinu (Colucci et al., 2017). Zadruhé vedla k identifikaci souvislosti mezi aktivací kostních a jaterních receptorů BMP typu I. Zatřetí tento případ posiluje význam intaktního TMPRSS6 v řízení jaterní signalizace BMP/SMAD, protože u jiných pacientů s FOP se stejnou mutací ACVR1 a pravděpodobně normálním TMPRSS6 nebyla identifikována žádná IRIDA (Pagani et al., 2017). A konečně, tento případ je v souladu s představou, že haploinsuficience TMPRSS6 nemůže způsobit klasickou IRIDA.
Optimální léčba IRIDA není definována. Perorální podávání železa je neúčinné, protože se nevstřebává. Přidání vitaminu C umožňuje sporadickou odpověď. Intravenózní železo vyvolává částečnou odpověď obvykle pomaleji ve srovnání s pacienty se získaným nedostatkem železa. EPO je v klasických případech neúčinné (De Falco et al., 2013; Heeney a Finberg, 2014).
Anémie adenomů produkujících hepcidin
Jedná se o extrémně vzácný stav u dospělých pacientů postižených glykogenovou střádavou chorobou 1a, což je recesivní porucha způsobená nedostatkem glukóza-6-fosfatázy, která katalyzuje reakci podílející se na glykogenolýze i glukoneogenezi. Častým nebezpečným příznakem onemocnění je hypoglykémie. Současná léčba vede k prodloužení přežití postižených dětí až do dospělého věku s výskytem několika komplikací, jako je anémie a jaterní adenomy. Anémie je mikrocytární a hypochromní, s nedostatkem železa a refrakterní na perorální léčbu železem. Anémie se vrací po chirurgické resekci adenomu. Ve tkáni adenomu byla zjištěna pozitivní mRNA hepcidinu, zatímco normální okolní tkáň vykazovala supresi hepcidinu, jak se očekávalo v důsledku ektopické nekontrolované produkce hepcidinu (Weinstein et al., 2002). Hematologické vlastnosti pacientů se podobají vlastnostem pacientů s IRIDA, protože mají společné vysoké hladiny hepcidinu jako společný mechanismus anémie.
Anémie zánětu
Anémie zánětu (AI), dříve známá jako anémie chronických onemocnění, je středně těžká normochromní-normocytární anémie, která se rozvíjí v podmínkách systémového zánětu a imunitní aktivace. Vyskytuje se u několika běžných onemocnění, včetně chronických infekcí, autoimunitních onemocnění, pokročilého nádorového onemocnění, chronického onemocnění ledvin, městnavého srdečního selhání, chronické obstrukční plicní nemoci, anémie starších osob (alespoň částečně) a onemocnění štěpu proti hostiteli. AI je jednou z nejčastějších anémií na světě a nejčastější anémií u hospitalizovaných pacientů. Akutní zánět přispívá k závažnosti anémie na jednotkách intenzivní péče. Molekulární mechanismy, které jsou základem AI, jsou mnohočetné a složité. Nadprodukce cytokinů, jako jsou IL1-β, TNF-α a IL-6 makrofágy a INF-γ lymfocyty, otupuje produkci EPO, zhoršuje odpověď na erytropoézu, zvyšuje hladinu hepcidinu a může aktivovat erytrofagocytózu, zejména u akutních forem (Weiss a Goodnough, 2005; Ganz, 2019).
Hepcidin je aktivován IL-6 prostřednictvím receptoru IL-6 (IL-6R) a signalizace JAK2-STAT3. Plná aktivace hepcidinu vyžaduje aktivní dráhu BMP-SMAD, protože inaktivace signalizace BMP snižuje hepcidin ve zvířecích modelech zánětu (Theurl et al., 2011). Deregulace systémové homeostázy železa způsobuje sekvestraci železa v makrofázích a sníženou absorpci a recyklaci, což vede k nízké saturaci transferinu a omezení erytropoézy a dalších tkání železem.
Tradiční léčba AI je založena na reverzibilitě/kontrole základního onemocnění, pokud je to možné. Pokud je onemocnění neléčitelné a anémie je mírná, je třeba pečlivě zhodnotit rizika a přínosy, aby se předešlo nežádoucím účinkům jakékoli léčby. Léčba založená na patofyziologii je omezena na sloučeniny podobné erytropoetinu a železo. Použití látek stimulujících erytropoézu (ESA) potlačuje hepcidin tím, že vyvolává expanzi erytropoézy. Tento přístup je široce používán u pacientů s chronickým onemocněním ledvin, myelodysplastickými syndromy s nízkým rizikem a nádorovými onemocněními podstupujícími chemoterapii. Je však nutná pečlivá klinická kontrola, protože vysoké dávky mají kardiovaskulární nežádoucí účinky. Podávání intravenózního železa může zmírnit restrikci železa způsobenou expanzí erytropoézy závislou na ESA. Perorální podávání železa je obvykle neúčinné, protože vysoké hladiny hepcidinu působí proti jeho střevní absorpci. U chronického onemocnění ledvin se experimentálně používají inhibitory prolylhydroxylázy (hypoxií indukovatelný faktor, stabilizátory HIF) s cílem zvýšit endogenní EPO. Chronická léčba transfuzí červených krvinek se nedoporučuje kvůli přechodnému účinku a nežádoucím účinkům; omezuje se na těžké refrakterní anémie (Camaschella, 2019; Weiss et al., 2019).
Anémie spojené s nízkou hladinou hepcidinu
Neúčinná erytropoéza a nízká nebo nepřiměřeně normální hladina hepcidinu s následným přetížením železem jsou typickými znaky tzv.“ Prototypem je β-talasemie, genetické recesivní onemocnění způsobené mutací β-globinového genu, která způsobuje anémii a nadměrnou produkci α-globinových řetězců. Ten se v kostní dřeni vysráží v podobě hemichromů, které poškozují dozrávající erytroidní prekurzory a vedou k neúčinné erytropoéze. K tomu dochází u netransfuzně závislé talasemie nebo talasemie intermedia, jejichž erytropoéza je charakterizována převahou nezralých buněk, které uvolňují erytroferon, aby inhibovaly expresi jaterního hepcidinu. Hladiny hepcidinu jsou obvykle vyšší u transfuzně závislé talasemie, kde je endogenní neefektivní erytropoéza alespoň částečně potlačena transfuzí (Camaschella a Nai, 2016).
Potlačení hepcidinu je zprostředkováno zvýšeným množstvím cytokinu erytroferronu (ERFE), člena rodiny TNF-α kódovaného genem ERFE, syntetizovaného erytroblasty při stimulaci EPO (Kautz et al., 2014). ERFE se uvolňuje do oběhu a sekvestruje BMP, zejména BMP6 (Arezes et al., 2018), čímž oslabuje signalizaci hepcidinu v reakci na železo. Kromě toho dochází k epigenetické supresi v lokusu hepcidinu prostřednictvím histonové deacetylázy HDAC3 (Pasricha et al., 2017). Když anémie způsobí hypoxii, působí další mediátory, jako je PDGF-BB (Sonnweber et al., 2014), který je uvolňován různými typy buněk, potlačují hepcidin.
Hladiny hepcidinu jsou sníženy zvláštním mechanismem u nízkorizikové myelodysplazie s prstencovitými sideroblasty, klonální poruchy způsobené mutacemi spliceosomového genu SF3B1. Železo se hromadí v mitochondriích, což vede k neúčinné erytropoéze a systémovému přetížení železem. Abnormálně sestřihaný, prodloužený protein ERFE je při potlačování hepcidinu silnější než divoký typ ERFE (Bondu et al., 2019) a způsobuje na transfuzi nezávislou zátěž železem.
Targeted Therapies for Hepcidin-Related Anemias
Identifikace molekulárních mechanismů zodpovědných za dříve diskutované anémie podnítila výzkum v oblasti vývoje cílené léčby, která by nahradila současnou symptomatickou léčbu (Sebastiani et al., 2016; Crielaard et al., 2017). Přístupy se liší podle typu anémie a cíle snížit či zvýšit hladinu hepcidinu nebo jeho účinek (tabulka 2).
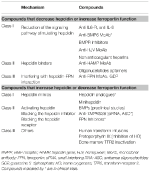
Tabulka 2. Experimentální terapie zaměřené na osu hepcidin-ferroportin.
Experimentální terapie zaměřené na snížení hladiny hepcidinu/zvýšení funkce ferroportinu
S výjimkou nádorů produkujících hepcidin, které musí být chirurgicky odstraněny, mohou být sloučeniny, které antagonizují hepcidin nebo jeho účinky, užitečné u všech anémií charakterizovaných vysokou hladinou hepcidinu. Jejich hlavní využití by bylo u chronických zánětlivých onemocnění s cílem zvrátit hypoferémii a anémii. V preklinických studiích bylo zkoumáno několik experimentálních terapií zaměřených na manipulaci s dráhou hepcidinu a jeho funkcí. Antagonisté hepcidinu jsou inhibitory syntézy/regulátorů hepcidinu (Ganz, 2019), látky vážící hepcidin, které blokují jeho funkci, a sloučeniny, které zasahují do interakce hepcidin-ferroportin (tabulka 2). Některé sloučeniny jsou v klinických studiích zejména u chronických onemocnění ledvin (Sheetz et al., 2019). U IRIDA byla v preklinických studiích navržena manipulace s dráhou hepcidinu pomocí anti-HJV MoAb (Kovac et al., 2016).
Experimentální terapie ke zvýšení hladiny hepcidinu/snížení funkce feroportinu
Zvýšení hladiny hepcidinu může nejen snížit přetížení železem, ale také částečně kontrolovat neefektivní erytropoézu u anémií zatížených železem. Z těchto stavů je nejvíce studována β-talasemie (Casu et al., 2018; Gupta et al., 2018). Navrhovanými léky jsou analoga hepcidinu (některá v klinických studiích), modulátory hepcidinu, zejména inhibitory TMPRSS6, nebo sloučeniny, které zasahují do interakce hepcidin-feroportin snižující export železa (tab. 2).
Zatímco sloučeniny zvyšující hepcidin snižují neefektivní erytropoézu v důsledku bludného kruhu mezi neefektivní erytropoézou a zátěží železem (Camaschella a Nai, 2016), léky, které podporují zrání erytroidních prekurzorů, jako je past na ligandy receptoru aktivinu IIB, luspatercept, nejen zlepšují anémii, ale také zlepšují homeostázu železa snížením inhibice hepcidinu (Piga et al., 2019).
Některé cílené přístupy, které jsou nyní v klinických studiích, snad povedou k novým způsobům léčby různých anémií.
Závěr
Velkolepý pokrok v pochopení regulace metabolismu železa a hepcidinu umožnil lépe porozumět řízení erytropoézy, protože železo je spolu s erytropoetinem základním faktorem zrání erytroidních buněk. Stavy, které vedou k anémii, mohou být spojeny s vysokou a nízkou hladinou hepcidinu. V obou případech může kontrastní deregulace hepcidinu zlepšit/upravit anémii v preklinických modelech a nabídnout nové nástroje, které jsou již nebo budou brzy klinicky zkoumány pro léčbu specifických anémií.
Příspěvky autorů
AP vypracoval návrh článku. CC vypracoval konečnou verzi. AN a LS se podíleli na sepsání a kritické revizi rukopisu. Všichni autoři schválili konečnou verzi.
Financování
Tento článek byl částečně podpořen grantem EHA pro pokročilý výzkum v roce 2018 pro AP. Cariplo Foundation Young Investigator Grant n° 2017-0916 pro AN.
Konflikt zájmů
CC je konzultantem společností Vifor Pharma, Celgene a Novartis.
Zbylí autoři prohlašují, že výzkum byl prováděn bez jakýchkoli komerčních nebo finančních vztahů, které by mohly být považovány za potenciální střet zájmů.
An, P., Wu, Q., Wang, H., Guan, Y., Mu, M., Liao, Y. a další (2012). Varianty TMPRSS6, ale ne TF, TFR2 nebo BMP2 jsou spojeny se zvýšeným rizikem anémie z nedostatku železa. Hum. Mol. Genet. 21, 2124-2131. doi: 10.1093/hmg/dds028
PubMed Abstract | CrossRef Full Text | Google Scholar
Andriopoulos, B. Jr, Corradini, E., Xia, Y., Faasse, S. A., Chen, S., Grgurevic, L., et al. (2009). BMP6 je klíčovým endogenním regulátorem exprese hepcidinu a metabolismu železa. Nat. Genet. 41, 482-487. doi: 10.1038/ng.335
PubMed Abstract | CrossRef Full Text | Google Scholar
Arezes, J., Foy, N., McHugh, K., Sawant, A., Quinkert, D., Terraube, V., et al. (2018). Erythroferron inhibuje indukci hepcidinu působením BMP6. Blood 132, 1473-1477. doi: 10.1182/blood-2018-06-857995
PubMed Abstract | CrossRef Full Text | Google Scholar
Aschemeyer, S., Qiao, B., Stefanova, D., Valore, E. V., Sek, A. C., Ruwe, T. A., et al. (2018). Strukturně-funkční analýza ferroportinu definuje vazebné místo a alternativní mechanismus účinku hepcidinu. Blood. 131, 899-910.doi: 10.1182/blood-2017-05-786590
CrossRef Full Text | Google Scholar
Babitt, J. L., Huang, F. W., Wrighting, D. M., Xia, Y., Sidis, Y., Samad, T. A., et al. (2006). Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat. Genet. 38, 531-539. doi: 10.1038/ng1777
PubMed Abstract | CrossRef Full Text | Google Scholar
Beguin, Y. (2003). Rozpustný transferinový receptor pro hodnocení erytropoézy a stavu železa. Clin. Chim. Acta 329, 9-22. doi: 10.1016/S0009-8981(03)00005-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Bondu, S., Alary, A. S., Lefevre, C., Houy, A., Jung, G., Lefebvre, T., et al. (2019). Varianta erytroferonu narušuje homeostázu železa u myelodysplastického syndromu s mutací SF3B1. Sci. Transl. Med. 11:pii:eaav5467. doi: 10.1126/scitranslmed.aav5467
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C. (2013). Jak vedu pacienty s atypickou mikrocytární anémií. Br. J. Haematol. 160, 12-24. doi: 10.1111/bjh.12081
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C. (2019). Nedostatek železa. Blood 133, 30-39. doi: 10.1182/blood-2018-05-815944
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C., and Nai, A. (2016). Neefektivní erytropoéza a regulace stavu železa u anémií se zátěží železem. Br. J. Haematol. 172, 512-523. doi: 10.1111/bjh.13820
PubMed Abstract | CrossRef Full Text | Google Scholar
Camaschella, C., and Pagani, A. (2018). Pokroky v chápání metabolismu železa a jeho křížení s erytropoézou. Br. J. Haematol. 182, 481-494. doi: 10.1111/bjh.15403
PubMed Abstract | CrossRef Full Text | Google Scholar
Canali, S., Wang, C. Y., Zumbrennen-Bullough, K. B., Bayer, A., and Babitt, J. L. (2017). Kostní morfogenetický protein 2 řídí homeostázu železa u myší nezávisle na Bmp6. Am. J. Hematol. 92, 1204-1213. doi: 10.1002/ajh.24888
PubMed Abstract | CrossRef Full Text | Google Scholar
Casu, C., Nemeth, E., and Rivella, S. (2018). Agonisté hepcidinu jako terapeutické nástroje. Blood 131, 1790-1794. doi: 10.1182/blood-2017-11-737411
PubMed Abstract | CrossRef Full Text | Google Scholar
Colucci, S., Pagani, A., Pettinato, M., Artuso, I., Nai, A., Camaschella, C. a další (2017). Imunofilin FKBP12 inhibuje expresi hepcidinu vazbou na receptor BMP typu I ALK2 v hepatocytech. Blood 130, 2111-2120. doi: 10.1182/blood-2017-04-780692
PubMed Abstract | CrossRef Full Text | Google Scholar
Crielaard, B. J., Lammers, T., and Rivella, S. (2017). Cílení na metabolismus železa při objevování a podávání léčiv. Nat. Rev. Drug Discov. 16, 400-423. doi: 10.1038/nrd.2016.248
PubMed Abstract | CrossRef Full Text | Google Scholar
De Falco, L., Sanchez, M., Silvestri, L., Kannengiesser, C., Muckenthaler, M. U., Iolascon, A., et al. (2013). Anémie z nedostatku železa refrakterní na železo. Haematologica 98, 845-853. doi: 10.3324/haematol.2012.075515
PubMed Abstract | CrossRef Full Text | Google Scholar
De Falco, L., Silvestri, L., Kannengiesser, C., Moran, E., Oudin, C., Rausa, M., et al. (2014). Funkční a klinický dopad nových variant TMPRSS6 u pacientů s anémií z nedostatku železa refrakterní na železo a genotypově-fenotypové studie. Hum. Mutat. 35, 1321-1329. doi: 10.1002/humu.22632
PubMed Abstract | CrossRef Full Text | Google Scholar
Donker, A. E., Schaap, C. C., Novotny, V. M., Smeets, R., Peters, T. M., van den Heuvel, B. L., et al. (2016). Iron refractory iron deficiency anemia: a heterogeneous disease that is not always iron refractory. Am. J. Hematol. 91, E482-E490. doi: 10.1002/ajh.24561
PubMed Abstract | CrossRef Full Text | Google Scholar
Du, X., She, E., Gelbart, T., Truksa, J., Lee, P., Xia, Y., et al. (2008). Serinová proteáza TMPRSS6 je nezbytná pro vnímání nedostatku železa. Science 320, 1088-1092. doi: 10.1126/science.1157121
PubMed Abstract | CrossRef Full Text | Google Scholar
Finberg, K. E., Heeney, M. M., Campagna, D. R., Aydinok, Y., Pearson, H. A., Hartman, K. R., et al. (2008). Mutace v TMPRSS6 způsobují anémii z nedostatku železa (IRIDA). Nat. Genet. 40, 569-571. doi: 10.1038/ng.130
PubMed Abstract | CrossRef Full Text | Google Scholar
Ganz, T. (2019). Zánětlivá anémie. N. Engl. J. Med. 381, 1148-1157. doi: 10.1056/NEJMra1804281
PubMed Abstract | CrossRef Full Text | Google Scholar
Gupta, R., Musallam, K. M., Taher, A. T., and Rivella, S. (2018). Neefektivní erytropoéza: anémie a přetížení železem. Hematol. Oncol. Clin. North Am. 32, 213-221. doi: 10.1016/j.hoc.2017.11.009
PubMed Abstract | CrossRef Full Text | Google Scholar
Heeney, M. M., and Finberg, K. E. (2014). Iron-refractory iron deficiency anemia (IRIDA) (anémie z nedostatku železa refrakterní na železo). Hematol. Oncol. Clin. North Am. 28, 637-652. doi: 10.1016/j.hoc.2014.04.009
PubMed Abstract | CrossRef Full Text | Google Scholar
Heeney, M. M., Guo, D., De Falco, L., Campagna, D. R., Olbina, G., Kao, P. P., et al. (2018). Normalizace hepcidinu předpovídá stav mutace TMPRSS6 u pacientů s chronickým nedostatkem železa. Blood 132, 448-452. doi: 10.1182/blood-2017-03-773028
PubMed Abstract | CrossRef Full Text | Google Scholar
Kautz, L., Jung, G., Valore, E. V., Rivella, S., Nemeth, E., and Ganz, T. (2014). Identifikace erythroferronu jako erytroidního regulátoru metabolismu železa. Nat. Genet. 46, 678-684. doi: 10.1038/ng.2996
PubMed Abstract | CrossRef Full Text | Google Scholar
Koch, P. S., Olsavszky, V., Ulbrich, F., Sticht, C., Demory, A., Leibing, T., et al. (2017). Angiocrinní signalizace Bmp2 v myších játrech řídí normální homeostázu železa. Blood 129, 415-419. doi: 10.1182/blood-2016-07-729822
PubMed Abstract | CrossRef Full Text | Google Scholar
Kovac, S., Boser, P., Cui, Y., Ferring-Appel, D., Casarrubea, D., Huang, L., et al. (2016). Protilátka proti hemojuvelinu koriguje anémii způsobenou nepřiměřeně vysokou hladinou hepcidinu. Haematologica 101, e173-e176. doi: 10.3324/haematol.2015.140772
PubMed Abstract | CrossRef Full Text | Google Scholar
Lakhal, S., Schodel, J., Townsend, A. R., Pugh, C. W., Ratcliffe, P. J., and Mole, D. R. (2011). Regulace transmembránové serinové proteinázy typu II TMPRSS6 hypoxií indukovanými faktory: nové spojení mezi signalizací hypoxie a homeostázou železa. J. Biol. Chem. 286, 4090-4097. doi: 10.1074/jbc.M110.173096
PubMed Abstract | CrossRef Full Text | Google Scholar
Meynard, D., Kautz, L., Darnaud, V., Canonne-Hergaux, F., Coppin, H., and Roth, M. P. (2009). Nedostatek kostního morfogenetického proteinu BMP6 vyvolává masivní přetížení železem. Nat. Genet. 41, 478-481. doi: 10.1038/ng.320
PubMed Abstract | CrossRef Full Text | Google Scholar
Muckenthaler, M. U., Rivella, S., Hentze, M. W., and Galy, B. (2017). Červený koberec pro metabolismus železa. Cell 168, 344-361. doi: 10.1016/j.cell.2016.12.034
PubMed Abstract | CrossRef Full Text | Google Scholar
Nemeth, E., Tuttle, M. S., Powelson, J., Vaughn, M. B., Donovan, A., Ward, D. M., et al. (2004). Hepcidin reguluje buněčný eflux železa vazbou na ferroportin a indukcí jeho internalizace. Science 306, 2090-2093. doi: 10.1126/science.1104742
PubMed Abstract | CrossRef Full Text | Google Scholar
Pagani, A., Colucci, S., Bocciardi, R., Bertamino, M., Dufour, C., Ravazzolo, R., et al. (2017). Nová forma IRIDA v důsledku kombinovaných heterozygotních mutací TMPRefSS6 a ACVR1A kódujících receptor BMP ALK2. Blood 129, 3392-3395. doi: 10.1182/blood-2017-03-773481
PubMed Abstract | Cross Full Text | Google Scholar
Pagani, A., Vieillevoye, M., Nai, A., Rausa, M., Ladli, M., Lacombe, C., et al. (2015). Regulace buněčného povrchového transferinového receptoru-2 štěpením závislým na železe a uvolňováním rozpustné formy. Haematologica 100, 458-465. doi: 10.3324/haematol.2014.118521
PubMed Abstract | CrossRef Full Text | Google Scholar
Pasricha, S. R., Lim, P. J., Duarte, T. L., Casu, C., Oosterhuis, D., Mleczko-Sanecka, K., et al. (2017). Hepcidin je regulován promotorovou acetylací histonů a HDAC3. Nat. Commun. 8:403. doi: 10.1038/s41467-017-00500-z
PubMed Abstract | CrossRef Full Text | Google Scholar
Piga, A., Perrotta, S., Gamberini, M. R., Voskaridou, E., Melpignano, A., Filosa, A., et al. (2019). Luspatercept zlepšuje hladiny hemoglobinu a požadavky na krevní transfuze ve studii pacientů s beta-talasemií. Blood 133, 1279-1289. doi: 10.1182/blood-2018-10-879247
PubMed Abstract | CrossRef Full Text | Google Scholar
Rausa, M., Pagani, A., Nai, A., Campanella, A., Gilberti, M. E., Apostoli, P., et al. (2015). Exprese Bmp6 v myších jaterních neparenchymových buňkách: mechanismus kontroly jejich vysoké aktivity exportéru železa a ochrany hepatocytů před přetížením železem? PLoS One 10:e0122696. doi: 10.1371/journal.pone.0122696
PubMed Abstract | CrossRef Full Text | Google Scholar
Sebastiani, G., Wilkinson, N., and Pantopoulos, K. (2016). Farmakologické cílení osy hepcidin/ferroportin. Front. Pharmacol. 7:160. doi: 10.3389/fphar.2016.00160
PubMed Abstract | CrossRef Full Text | Google Scholar
Sheetz, M., Barrington, P., Callies, S., Berg, P. H., McColm, J., Marbury, T., et al. (2019). Cílení na dráhu hepcidin-ferroportin u anémie při chronickém onemocnění ledvin. Br. J. Clin. Pharmacol. 85, 935-948. doi: 10.1111/bcp.13877
PubMed Abstract | CrossRef Full Text | Google Scholar
Shore, E. M., Xu, M., Feldman, G. J., Fenstermacher, D. A., Cho, T. J., Choi, I. H., et al. (2006). Opakující se mutace v receptoru BMP typu I ACVR1 způsobuje dědičnou a sporadickou fibrodysplasia ossificans progressiva. Nat. Genet. 38, 525-527. doi: 10.1038/ng1783
PubMed Abstract | CrossRef Full Text | Google Scholar
Silvestri, L., Pagani, A., Nai, A., De Domenico, I., Kaplan, J., and Camaschella, C. (2008). Serinová proteáza matriptáza-2 (TMPRSS6) inhibuje aktivaci hepcidinu štěpením membránového hemojuvelinu. Cell Metab. 8, 502-511. doi: 10.1016/j.cmet.2008.09.012
PubMed Abstract | CrossRef Full Text | Google Scholar
Silvestri, L., Rausa, M., Pagani, A., Nai, A., and Camaschella, C. (2013). Jak posoudit kauzalitu mutací TMPRSS6? Hum. Mutat. 34, 1043-1045. doi: 10.1002/humu.22321
PubMed Abstract | CrossRef Full Text | Google Scholar
Sonnweber, T., Nachbaur, D., Schroll, A., Nairz, M., Seifert, M., Demetz, E. a další (2014). Hypoxií indukovaná downregulace hepcidinu je zprostředkována destičkovým růstovým faktorem BB. Gut 63, 1951-1959. doi: 10.1136/gutjnl-2013-305317
PubMed Abstract | CrossRef Full Text | Google Scholar
Sorensen, E., Rigas, A. S., Didriksen, M., Burgdorf, K. S., Thorner, L. W., Pedersen, O. B., et al. (2019). Genetické faktory ovlivňující hladinu hemoglobinu u 15 567 dárců krve: výsledky dánské studie dárců krve. Transfusion 59, 226-231. doi: 10.1111/trf.15075
CrossRef Full Text | Google Scholar
Theurl, I., Schroll, A., Sonnweber, T., Nairz, M., Theurl, M., Willenbacher, W., et al. (2011). Farmakologická inhibice exprese hepcidinu zvrátí anémii chronického zánětu u potkanů. Blood 118, 4977-4984. doi: 10.1182/blood-2011-03-345066
PubMed Abstract | CrossRef Full Text | Google Scholar
Weinstein, D. A., Roy, C. N., Fleming, M. D., Loda, M. F., Wolfsdorf, J. I., and Andrews, N. C. (2002). Nevhodná exprese hepcidinu je spojena s anémií refrakterní na železo: důsledky pro anémii chronických onemocnění. Blood 100, 3776-3781. doi: 10.1182/blood-2002-04-1260
PubMed Abstract | CrossRef Full Text | Google Scholar
Weiss, G., Ganz, T., and Goodnough, L. T. (2019). Zánětlivá anémie. Blood 133, 40-50. doi: 10.1182/blood-2018-06-856500
PubMed Abstract | CrossRef Full Text | Google Scholar
Weiss, G., and Goodnough, L. T. (2005). Anémie chronických onemocnění. N. Engl. J. Med. 352, 1011-1023. doi: 10.1056/NEJMra041809
PubMed Abstract | CrossRef Full Text | Google Scholar
.
Leave a Reply