Endocrinopatie: Il nuovo prezzo del trattamento del cancro
Gli inibitori del checkpoint immunitario (ICI) sono anticorpi monoclonali che bloccano i checkpoint – cioè l’antigene 4 dei linfociti T citotossici (CTLA-4), la proteina 1 della morte cellulare programmata (PD-1) e la proteina 1 della morte cellulare programmata (PD-L1) – portando così alla de-repressione della funzione delle cellule T citotossiche e a una migliore risposta immunitaria antitumorale (Figura 1).

Figura 1. Meccanismi degli inibitori del checkpoint immunitario.
Nel 2011, la US Food and Drug Administration ha approvato il primo ICI, ipilimumab, per l’uso nel melanoma. Da allora, cinque ICI sono stati approvati per la pratica clinica, compresi gli inibitori PD-1 (pembrolizumab e nivolumab) e gli inibitori PD-L1 (atezolizumab, avelumab e durvalumab).
Le ICI sono emerse come una nuova causa di eventi avversi endocrini immuno-correlati (IRAEs) che colpiscono l’ipofisi, la tiroide, il pancreas endocrino e (raramente) le ghiandole surrenali e paratiroidi. Il meccanismo alla base di queste endocrinopatie sembra essere mediato dal sistema immunitario, soprattutto dalle cellule T; tuttavia, i meccanismi specifici devono ancora essere chiariti e sono oggetto di studi recenti e in corso.
L’aumento dell’uso degli ICI, che sono molto efficaci contro vari tumori maligni, probabilmente aumenterà il carico della popolazione di queste endocrinopatie. Quindi, è essenziale per i medici essere in grado di identificarle e iniziare la loro gestione.
Monitoraggio delle endocrinopatie
Prima di iniziare gli ICI, i pazienti dovrebbero sottoporsi a test di laboratorio che includono TSH sierico, tiroxina libera sierica, cortisolo sierico 8 AM, ACTH sierico, e glucosio plasmatico a digiuno. Se uno di questi test è anormale, possono essere eseguiti ulteriori test e monitoraggi. I test di laboratorio specifici dovrebbero essere eseguiti anche quando ci sono sintomi o segni di qualsiasi endocrinopatia, come sarà discusso di seguito.
Il medico dovrebbe avere una soglia bassa per testare per l’endocrinopatia se il paziente ha una storia personale o familiare di autoimmunità o ha sviluppato altre IRAE. Sulla base della nostra esperienza clinica e delle linee guida del National Comprehensive Cancer Network, potrebbe essere necessario interrompere temporaneamente l’ICI di partenza se un paziente presenta ipofisite con effetti di massa, crisi surrenale, tireotossicosi grave o chetoacidosi diabetica.
Ipofisite
L’ipofisite è l’endocrinopatia più comune associata all’inibitore CTLA-4 ipilimumab, che si verifica nell’8%-11% dei pazienti. Si osserva meno frequentemente con gli inibitori PD-1 e PD-L1 (0%-5%). Anche se l’ipofisite di solito si verifica 6-12 settimane dopo l’inizio degli ICI, alcuni casi sono stati riportati dopo una durata prolungata della terapia.
Presentazione e gestione
L’ipofisite può presentarsi con effetti di massa dovuti all’allargamento dell’ipofisi, con mal di testa, visione doppia e difetti del campo visivo. Che ci siano effetti di massa o meno, tutti i casi hanno carenze ormonali ipofisarie – più comunemente insufficienza surrenalica secondaria, che può presentarsi come un’emergenza di “crisi surrenalica”. Può anche causare ipotiroidismo centrale e talvolta ipogonadismo secondario.
Sintomi come affaticamento, debolezza, nausea, confusione, perdita di memoria, perdita della libido, anoressia, allucinazioni, intolleranza alla temperatura e sensazione soggettiva di febbre e brividi possono verificarsi. Questi sintomi sono spesso aspecifici e possono sovrapporsi ai sintomi costituzionali legati al cancro.
Pertanto, il medico deve avere un alto sospetto quando valuta l’ipofisite, soprattutto perché l’insufficienza surrenalica secondaria che la accompagna può essere pericolosa per la vita se non riconosciuta e gestita prontamente. L’ipofisi posteriore è raramente coinvolta, quindi il diabete insipido centrale è raro nel contesto dell’uso di ICI.
I test diagnostici dovrebbero includere la risonanza magnetica del cervello (Figura 2) e testare gli ormoni prodotti dall’ipofisi e dalle ghiandole bersaglio.
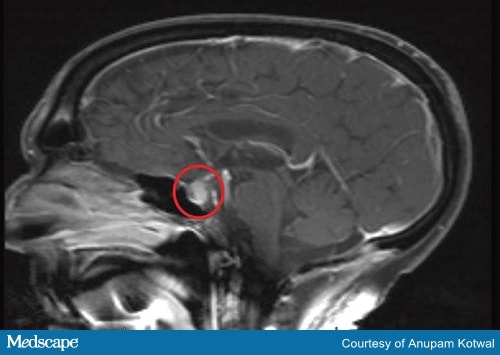
Figura 2a. Risonanza magnetica della testa, dimostrando l’allargamento e l’aumento della ghiandola pituitaria, con ispessimento del gambo.
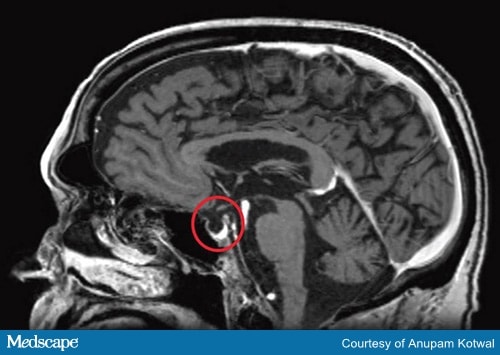
Figura 2b. Risonanza magnetica della testa, dimostrando la distruzione della ghiandola pituitaria, che appare come una sella vuota.
Oltre alle anomalie ormonali, anomalie elettrolitiche come l’iponatriemia e l’ipoglicemia possono verificarsi a causa di una carenza di cortisolo e tiroxina.
Quando un paziente presenta effetti di massa come mal di testa o anomalie della vista, l’ospedalizzazione dovrebbe essere considerata. Glucocorticoidi ad alte dosi (ad esempio, prednisone/metilprednisolone 1 mg/kg/die o desametasone 4 mg ogni 6 ore) devono essere somministrati non appena si teme che ci siano effetti di massa da ipofisite.
I pazienti che presentano ipotensione, confusione e ipoglicemia possono essere in crisi surrenalica, che deve essere gestita con una dose da sforzo di glucocorticoidi e rianimazione di liquidi. Una volta che la crisi surrenalica e gli effetti di massa si risolvono, o se il paziente presenta solo sintomi lievi o moderati di carenze ormonali, la sostituzione ormonale è il pilastro della gestione. La maggior parte dei pazienti che sviluppano ipopituitarismo richiedono una sostituzione ormonale per tutta la vita.
Tiroidite
La disfunzione tiroidea è l’endocrinopatia più comune associata agli inibitori PD-1 e PD-L1 (6%-21%). Si verifica meno comunemente con l’inibitore CTLA-4 ipilimumab. La terapia di combinazione con ipilimumab e un inibitore PD-1 ha dimostrato i più alti tassi di disfunzione tiroidea, che di solito si verifica 8-10 settimane dopo l’inizio di un ICI ma può essere ritardata fino a 2 anni.
Presentazione e gestione
La tiroidite può presentarsi con ipotiroidismo primario, talvolta preceduto da una fase tireotossica durante la quale vengono rilasciati ormoni tiroidei. I pazienti con preesistente tiroidite di Hashimoto possono avere un peggioramento dell’ipotiroidismo, suggerito da un aumento del dosaggio di sostituzione degli ormoni tiroidei.
Sembra esserci un rischio maggiore di progressione verso l’ipotiroidismo permanente nella tiroidite indotta da ICI rispetto alla tiroidite indolore, e questo è particolarmente vero quando gli anticorpi della perossidasi tiroidea (TPO) sono elevati. L’ipertiroidismo persistente, come la malattia di Graves, è stato riportato raramente con l’uso di inibitori CTLA-4.
Molti pazienti sono asintomatici a causa della natura acuta o lieve della disfunzione ormonale tiroidea, evidenziando l’importanza di monitorare i livelli di ormone tiroideo prima di ogni infusione di ICI. La maggior parte dei pazienti che presentano o progrediscono verso un ipotiroidismo manifesto richiedono una sostituzione a lungo termine dell’ormone tiroideo.
La diagnosi può essere facilmente stabilita utilizzando i test di funzionalità tiroidea. Gli anticorpi del recettore TSH devono essere testati solo quando l’ipertiroidismo è persistente per più di 8 settimane, nel qual caso un test positivo degli anticorpi del recettore TSH suggerirebbe la malattia di Graves.
L’imaging di solito non è necessario per fare una diagnosi di tiroidite indotta da ICI. In alcuni casi, l’ecografia tiroidea può aiutare a confermare la diagnosi di tiroidite, con la tiroide che appare eterogenea e ipovascolare (Figura 3).
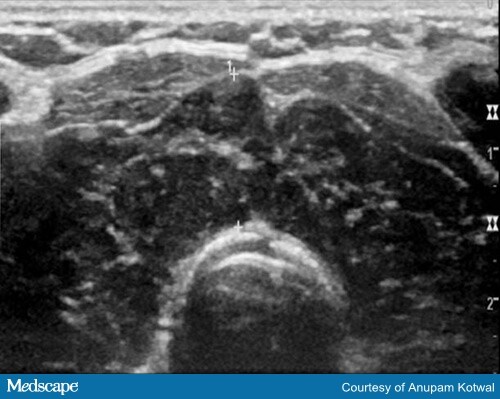
Figura 3. Immagine ecografica che dimostra un parenchima eterogeneo e ipoecogeno della ghiandola tiroidea, che è anche ipovascolare.
FDG-PET scansioni eseguite come parte di un workup di malignità o monitoraggio di solito dimostrano diffusamente aumentato assorbimento FDG nella tiroide, coerente con tiroidite (Figura 4).

Figura 4. Scansione FDG-PET che dimostra un aumento diffuso dell’assorbimento di FDG nella tiroide.
La gestione dipende dalla fase della tiroidite durante la quale viene fatta la diagnosi. La tireotossicosi transitoria può di solito essere gestita sintomaticamente con beta-bloccanti. Se c’è un disagio significativo al collo, che è raro, i glucocorticoidi possono essere considerati, ma di solito non sono necessari.
Per l’ipotiroidismo manifesto o l’ipotiroidismo subclinico sintomatico, la terapia sostitutiva con levotiroxina T4 dovrebbe essere iniziata e la sua dose titolata sulla base del TSH.
Diabete mellito insulino-dipendente
Nuova insorgenza di diabete mellito non è stata riportata negli studi clinici degli inibitori CTLA-4 ed è stata riportata in < 1% dei pazienti negli studi sugli inibitori PD-1. Tuttavia, tassi fino all’1,5% sono stati recentemente riportati con l’uso combinato di inibitori CTLA-4 e PD-1.
È stato dimostrato che gli inibitori PD-L1 non solo portano a un nuovo diabete insulino-dipendente, ma peggiorano anche il diabete preesistente. Gli alti titoli di anticorpi associati al diabete di tipo 1, riscontrati frequentemente, sono in linea con il processo immunitario.
Il diabete insulino-dipendente di nuova insorgenza si verifica di solito 20 settimane dopo l’inizio della terapia con inibitori PD-1, ma è stato riportato già 2 settimane e fino a 3-4 anni dopo l’inizio del trattamento.
Presentazione e gestione
A seconda della gravità della carenza di insulina, l’emergenza da chetoacidosi diabetica può verificarsi ed è stata riportata abbastanza frequentemente.
Si raccomanda che i pazienti con ICI che si presentano con iperglicemia siano testati per l’emoglobina A1c (di solito non estremamente elevata a causa della natura acuta dell’iperglicemia), C-peptide (di solito basso) con glucosio plasmatico concomitante, e anticorpi del diabete di tipo 1. Quando c’è qualche preoccupazione per la chetoacidosi diabetica, il bicarbonato sierico, il gap anionico, il beta-idrossibutirrato e i chetoni urinari dovrebbero essere testati. I medici dovrebbero avere una soglia bassa per gestire questi pazienti come quelli che hanno il diabete insulino-deficiente alla presentazione iniziale.
Una volta che l’episodio iniziale di iperglicemia si è risolto – e se gli anticorpi sono negativi e i livelli di C-peptide suggeriscono una produzione appropriata di insulina, soprattutto in assenza di iperglicemia preprandiale – può essere considerata una cauta de-escalation della terapia, ma i pazienti devono essere monitorati molto attentamente perché possono essere nella “fase della luna di miele” della carenza di insulina.
La maggior parte dei pazienti con diabete indotto da ICI richiede una terapia insulinica intensiva con un regime basale-bolus o una pompa di insulina al follow-up, suggerendo che la remissione è improbabile. Questo aggiunge significativamente alla morbilità dei pazienti che stanno già affrontando il cancro.
Endocrinopatie rare: Adrenalite e Ipoparatiroidismo
Una presentazione clinica suggestiva di insufficienza surrenalica primaria è stata descritta in alcuni casi, ma di solito è difficile da differenziare dall’insufficienza surrenalica secondaria dovuta a ipofisite o all’uso cronico di glucocorticoidi (per il cancro o la gestione di altre IRAE).
Recentemente, due casi di ipocalcemia e basso ormone paratiroideo suggestivi di ipoparatiroidismo immunomediato sono stati riportati con l’uso di inibitori PD-1, uno dei quali mostrava anticorpi contro il recettore del calcio-sensing.
Segui Medscape su Facebook, Twitter, Instagram e YouTube
.
Leave a Reply