Endocrinopathies : Le nouveau prix du traitement du cancer
Les inhibiteurs de points de contrôle immunitaires (ICI) sont des anticorps monoclonaux qui bloquent les points de contrôle – à savoir l’antigène 4 des lymphocytes T cytotoxiques (CTLA-4), la protéine de mort cellulaire programmée 1 (PD-1) et le ligand 1 de la protéine de mort cellulaire programmée (PD-L1) – entraînant ainsi une dé-répression de la fonction des cellules T cytotoxiques et une réponse immunitaire antitumorale renforcée (figure 1).

Figure 1. Mécanismes des inhibiteurs de points de contrôle immunitaires.
En 2011, la Food and Drug Administration américaine a approuvé le premier ICI, l’ipilimumab, pour une utilisation dans le mélanome. Depuis, cinq ICI ont été approuvés pour la pratique clinique, notamment les inhibiteurs PD-1 (pembrolizumab et nivolumab) et les inhibiteurs PD-L1 (atezolizumab, avelumab et durvalumab).
Les ICI sont apparus comme une nouvelle cause d’effets indésirables endocriniens liés à l’immunité (IRAE) affectant l’hypophyse, la thyroïde, le pancréas endocrine et (rarement) les glandes surrénales et parathyroïdes. Le mécanisme sous-jacent de ces endocrinopathies semble être à médiation immunitaire, principalement par les cellules T ; cependant, les mécanismes spécifiques doivent encore être élucidés et font l’objet d’études récentes et en cours.
L’utilisation accrue des ICI, qui sont très efficaces contre diverses tumeurs malignes, est susceptible d’augmenter le fardeau démographique de ces endocrinopathies. Par conséquent, il est essentiel que les cliniciens soient en mesure de les identifier et d’initier leur prise en charge.
Surveillance des endocrinopathies
Avant l’initiation des ICIs, les patients doivent subir des tests de laboratoire comprenant la TSH sérique, la thyroxine libre sérique, le cortisol 8 AM sérique, l’ACTH sérique et la glycémie à jeun. Si l’un de ces tests est anormal, d’autres tests et une surveillance peuvent être effectués. Des tests de laboratoire spécifiques doivent également être effectués en présence de symptômes ou de signes d’une quelconque endocrinopathie, comme nous le verrons plus loin.
Le clinicien doit avoir un seuil bas pour tester une endocrinopathie si le patient a des antécédents personnels ou familiaux d’auto-immunité ou a développé d’autres IRAE. Sur la base de notre expérience clinique et des directives du National Comprehensive Cancer Network, il peut être nécessaire d’interrompre temporairement l’ICI incitatif si un patient présente une hypophysite avec effets de masse, une crise surrénalienne, une thyrotoxicose sévère ou une acidocétose diabétique.
Hypophysite
L’hypophysite est l’endocrinopathie la plus fréquente associée à l’ipilimumab, un inhibiteur de CTLA-4, et survient chez 8 % à 11 % des patients. Elle est moins fréquente avec les inhibiteurs de PD-1 et de PD-L1 (0%-5%). Bien que l’hypophysite survienne généralement 6 à 12 semaines après l’initiation des ICIs, certains cas ont été rapportés après une durée prolongée de traitement.
Présentation et prise en charge
L’hypophysite peut se présenter avec des effets de masse dus à une hypertrophie de l’hypophyse, avec des céphalées, une vision double et des troubles du champ visuel. Qu’il y ait ou non des effets de masse, tous les cas présentent des déficiences hormonales hypophysaires – le plus souvent une insuffisance surrénalienne secondaire, qui peut se présenter comme une urgence de « crise surrénalienne ». Elle peut également provoquer une hypothyroïdie centrale et parfois un hypogonadisme secondaire.
Des symptômes comme la fatigue, la faiblesse, les nausées, la confusion, les pertes de mémoire, la perte de libido, l’anorexie, les hallucinations, l’intolérance à la température et la sensation subjective de fièvre et de frissons peuvent apparaître. Ces symptômes sont souvent non spécifiques et peuvent se superposer aux symptômes constitutionnels liés au cancer.
Par conséquent, le clinicien doit avoir une forte suspicion lorsqu’il évalue une hypophysite, en particulier parce que l’insuffisance surrénale secondaire qui l’accompagne peut mettre en danger la vie du patient si elle n’est pas reconnue et prise en charge rapidement. L’hypophyse postérieure est rarement touchée, c’est pourquoi le diabète insipide central est rare dans le cadre de l’utilisation des ICI.
Les tests diagnostiques doivent inclure une IRM du cerveau (Figure 2) et des tests sur les hormones produites par l’hypophyse et les glandes cibles.
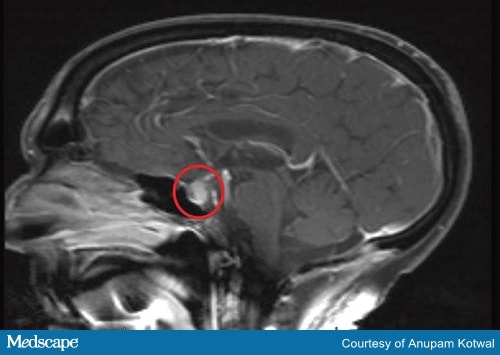
Figure 2a. IRM de la tête, démontrant l’élargissement et le rehaussement de l’hypophyse, avec un épaississement de la tige.
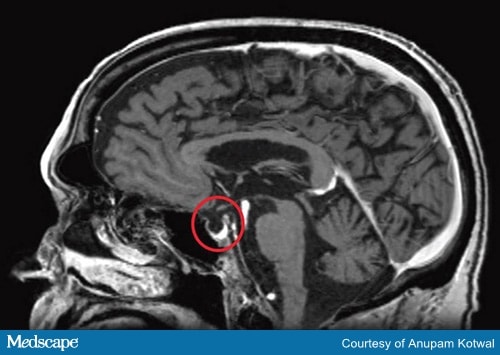
Figure 2b. IRM de la tête, démontrant la destruction de l’hypophyse, apparaissant comme une sella vide.
En plus des anomalies hormonales, des anomalies électrolytiques comme l’hyponatrémie et l’hypoglycémie peuvent survenir en raison d’une carence en cortisol et en thyroxine.
Lorsqu’un patient présente des effets de masse comme des céphalées ou des anomalies de la vision, une hospitalisation doit être envisagée. De fortes doses de glucocorticoïdes (par exemple, prednisone/méthylprednisolone 1 mg/kg/jour ou dexaméthasone 4 mg toutes les 6 heures) doivent être administrées dès que l’on craint des effets de masse dus à une hypophysite.
Les patients présentant une hypotension, une confusion et une hypoglycémie peuvent être en crise surrénalienne, qui doit être gérée avec une dose d’effort de glucocorticoïdes et une réanimation liquidienne. Une fois la crise surrénalienne et les effets de masse résolus, ou si le patient ne présente que des symptômes légers à modérés de déficiences hormonales, le remplacement hormonal est le pilier de la gestion. La plupart des patients qui développent un hypopituitarisme nécessitent un remplacement hormonal à vie.
Thyroïdite
La dysfonction thyroïdienne est l’endocrinopathie la plus fréquente associée aux inhibiteurs PD-1 et PD-L1 (6 %-21 %). Elle survient moins fréquemment avec l’ipilimumab, un inhibiteur de CTLA-4. Le traitement combiné avec l’ipilimumab et un inhibiteur de PD-1 a démontré les taux les plus élevés de dysfonctionnement thyroïdien, qui survient généralement 8 à 10 semaines après l’initiation d’un ICI mais peut être retardé jusqu’à 2 ans.
Présentation et prise en charge
La thyroïdite peut se présenter sous la forme d’une hypothyroïdie primaire, parfois précédée d’une phase thyrotoxique au cours de laquelle les hormones thyroïdiennes sont libérées. Les patients ayant une thyroïdite de Hashimoto préexistante peuvent présenter une aggravation de l’hypothyroïdie, suggérée par une augmentation de la posologie des hormones thyroïdiennes de substitution.
Il semble y avoir un risque plus élevé de progression vers une hypothyroïdie permanente dans les thyroïdites induites par les ICI par rapport aux thyroïdites indolores, et ceci est particulièrement vrai lorsque les anticorps anti-peroxydase thyroïdienne (TPO) sont élevés. Une hyperthyroïdie persistante, telle que la maladie de Graves, a rarement été rapportée avec l’utilisation des inhibiteurs de CTLA-4.
De nombreux patients sont asymptomatiques en raison de la nature aiguë ou légère du dysfonctionnement des hormones thyroïdiennes, soulignant l’importance de la surveillance des taux d’hormones thyroïdiennes avant chaque perfusion d’ICI. La plupart des patients qui présentent ou évoluent vers une hypothyroïdie manifeste nécessitent un remplacement à long terme des hormones thyroïdiennes.
Le diagnostic peut être facilement établi à l’aide de tests de la fonction thyroïdienne. Les anticorps anti-récepteurs de la TSH ne doivent être testés que lorsque l’hyperthyroïdie est persistante pendant plus de 8 semaines, auquel cas un test positif des anticorps anti-récepteurs de la TSH suggérerait une maladie de Basedow.
L’imagerie n’est généralement pas nécessaire pour établir un diagnostic de thyroïdite induite par l’ICI. Dans certains cas, l’échographie de la thyroïde peut aider à confirmer le diagnostic de thyroïdite, la thyroïde apparaissant hétérogène et hypovasculaire (figure 3).
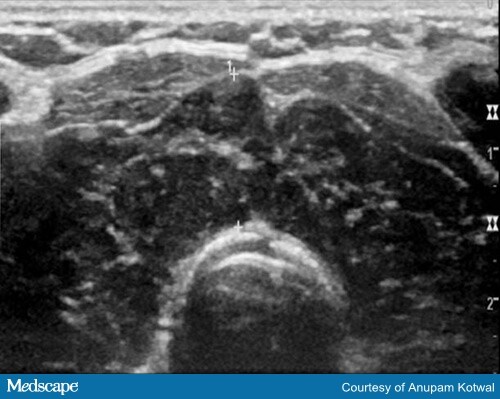
Figure 3. Image échographique montrant un parenchyme thyroïdien hétérogène et hypoéchogène, qui est également hypovasculaire.
La TEP-FDG réalisée dans le cadre d’un bilan de malignité ou d’un suivi montre généralement une augmentation diffuse de la captation du FDG dans la thyroïde, ce qui est cohérent avec une thyroïdite (figure 4).

Figure 4. Tomographie par TEP-FDG démontrant une augmentation diffuse de la captation de FDG dans la thyroïde.
La prise en charge dépend de la phase de la thyroïdite au cours de laquelle le diagnostic est posé. Une thyrotoxicose transitoire peut généralement être gérée de manière symptomatique avec des bêta-bloquants. En cas de gêne importante au niveau du cou, ce qui est rare, les glucocorticoïdes peuvent être envisagés, mais ils ne sont généralement pas nécessaires.
En cas d’hypothyroïdie manifeste ou d’hypothyroïdie subclinique symptomatique, un traitement de substitution de la T4 par la lévothyroxine doit être initié et sa dose titrée en fonction de la TSH.
Diabète insulino-dépendant
Une nouvelle apparition du diabète sucré n’a pas été rapportée dans les essais cliniques des inhibiteurs de CTLA-4 et a été rapportée chez < 1% des patients dans les études sur les inhibiteurs de PD-1. Cependant, des taux allant jusqu’à 1,5 % ont été récemment rapportés avec l’utilisation combinée des inhibiteurs de CTLA-4 et de PD-1.
Il a été démontré que les inhibiteurs de PD-L1 n’entraînent pas seulement l’apparition d’un nouveau diabète insulino-dépendant mais aggravent également le diabète préexistant. Les titres élevés d’anticorps associés au diabète de type 1 fréquemment rencontrés sont en accord avec le processus immunitaire.
Le diabète insulinodépendant d’apparition récente survient généralement 20 semaines après le début du traitement par inhibiteur de PD-1 mais a été rapporté aussi tôt que 2 semaines et aussi tard que 3-4 ans après le début du traitement.
Présentation et prise en charge
Selon la sévérité de la carence en insuline, l’urgence d’une acidocétose diabétique peut survenir et a été rapportée assez fréquemment.
Il est recommandé que les patients sous ICI présentant une hyperglycémie soient testés pour l’hémoglobine A1c (généralement pas extrêmement élevée en raison de la nature aiguë de l’hyperglycémie), le peptide C (généralement faible) avec le glucose plasmatique concomitant, et les anticorps du diabète de type 1. En cas de risque d’acidocétose diabétique, il convient de tester le bicarbonate sérique, le trou anionique, le bêta-hydroxybutyrate et les corps cétoniques urinaires. Les cliniciens doivent avoir un seuil bas pour prendre en charge ces patients comme ceux ayant un diabète insulinodéficitaire lors de la présentation initiale.
Une fois que l’épisode initial d’hyperglycémie s’est résolu – et si les anticorps sont négatifs et que les taux de peptide C suggèrent une production d’insuline appropriée, en particulier en l’absence d’hyperglycémie préprandiale – une désescalade prudente du traitement peut être envisagée, mais les patients doivent être surveillés très étroitement car ils peuvent être dans la « phase de lune de miel » de l’insulinodéficience.
La plupart des patients atteints de diabète induit par les ICI nécessitent une insulinothérapie intensive avec un régime basal-bolus ou une pompe à insuline lors du suivi, ce qui suggère que la rémission est peu probable. Cela ajoute considérablement à la morbidité des patients qui sont déjà confrontés à un cancer.
Endocrinopathies rares : Surrénalite et hypoparathyroïdie
Une présentation clinique évoquant une insuffisance surrénalienne primaire a été décrite dans certains rapports de cas, mais il est généralement difficile de la différencier d’une insuffisance surrénalienne secondaire due à une hypophysite ou à l’utilisation chronique de glucocorticoïdes (pour le cancer ou la gestion d’autres IRAE).
Récemment, deux cas d’hypocalcémie et d’hormone parathyroïdienne basse évoquant une hypoparathyroïdie à médiation immunitaire ont été rapportés avec l’utilisation d’inhibiteurs PD-1, l’un d’entre eux présentant des anticorps contre le récepteur de détection du calcium.
Suivez Medscape sur Facebook, Twitter, Instagram et YouTube
.
Leave a Reply